ΕΙΣΑΓΩΓΗ
Ο καρκίνος του μαστού είναι μία από τις κύριες αιτίες θανάτου από καρκίνο στις γυναίκες. Έχει αποδειχθεί ότι αποτελεί ετερογενή νόσο που περιλαμβάνει τουλάχιστον πέντε υποτύπους. Πολλαπλές μελέτες έχουν δείξει ότι αυτή η ετερογένεια είναι κυρίως αποτέλεσμα τριών παραγόντων: της έκφρασης οιστρογονικών υποδοχέων, του πολλαπλασιασμού των καρκινικών κυττάρων και της ενίσχυσης του γονιδίου ERBB2/HER2/neu. Οι δύο πιο σημαντικοί υπότυποι καρκίνου του μαστού διακρίνονται από την έκφραση ή όχι οιστρογονικών υποδοχέων. Οι όγκοι που εκφράζουν τους υποδοχείς ER αποδίδονται με τον όρο “luminal” και αντιπροσωπεύουν περίπου το 70%. Ο όρος αυτός υποδεικνύει την προέλευση του όγκου από τα αυλικά κύτταρα που επενδύουν τους πόρους του μαζικού αδένα. Η υποομάδα αυτή των όγκων υποδιαρείται σε “luminal A” και “ luminal Β” ανάλογα με την έκφραση HER2 (θετική στο luminal B) και το ποσοστό έκφρασης του μιτωτικού δείκτη Κi67 (υψηλός στο luminal B).

Από τους
Παναγιώτα Οικονομοπούλου, Αθανάσιο Κωτσάκη και Νικόλαο Κεντεποζίδη
Οι αντιοιστρογονικές θεραπείες στοχεύουν στη μείωση των επιπέδων οιστρογόνων ή στην αναστολή της μετάδοσης σημάτων μέσω οιστρογονικών υποδοχέων. Υπάρχουν τρεις κατηγορίες αντιοιστρογονικών παραγόντων (Πίνακας 1): α) οι εκλεκτικοί τροποποιητές των οιστρογονικών υποδοχέων (Selective Estrogen Receptor Modulators-SERMs, όπως η ταμοξιφαίνη), που εμποδίζουν τη δραστηριότητα του υποδοχέα ER, β) οι αναστολείς σύνθεσης οιστρογόνων, (αναστολείς αρωματάσης, όπως η αναστραζόλη, η λετροζόλη και η εξαμεστάνη) και γ) οι εκλεκτικοί καταστολείς του οιστρογονικού υποδοχέα (Selective Estrogen Downregulators-SERDs, όπως η φουλβεστράνη) που επάγουν την αποσταθεροποίηση και αποδόμηση του υποδοχέα.
| Ορμονοθεραπεία | Μηχανισμός | Παραδείγματα |
| Εκλεκτικοί τροποποιητές των οιστρογονικών υποδοχέων (Selective Estrogen Receptor Modulators-SERMs) | Αναστολείς του οιστρογονικού υποδοχέα, μικτή ανταγωνιστική και αγωνιστική δράση | Ταμοξιφαίνη, ραλοξιφαίνη |
| Εκλεκτικοί καταστολείς των οιστρογονικών υποδοχέων (Selective Estrogen Downregulators-SERDs) | Προωθούν την αποδόμηση του οιστρογονικού υποδοχέα | Φουλβεστράνη |
| Αναστολείς αρωματάσης | Εμποδίζουν τη σύνθεση των οιστρογόνων | Λετροζόλη, αναστραζόλη, εξαμεστάνη |
Πίνακας 1. Είδη και μηχανισμός δράσης ορμονοθεραπείας.
Τα τελευταία χρόνια, ο αντιοιστρογονικός παράγοντας ταμοξιφαίνη αποτελεί τη βασική θεραπεία για ασθενείς με καρκίνο του μαστού που εκφράζουν οιστρογονικούς υποδοχείς, τόσο στην πρώιμη όσο και στην προχωρημένη νόσο. Παρόλα αυτά, σχεδόν 50% των ασθενών με μεταστατική νόσο δεν ανταποκρίνονται στη θεραπεία πρώτης γραμμής με ταμοξιφαίνη (πρωτογενής αντίσταση-de novo resistance). Επιπλέον, ένα μεγάλο ποσοστό ασθενών υποτροπιάζουν μετά τη θεραπεία με ταμοξιφαίνη, παρά την αρχική ανταπόκριση (επίκτητη αντίσταση-acquired resistance). Αυτή η διαφορετική ανταπόκριση μπορεί να πηγάζει από την έκφραση συγκεκριμένων παραγόντων που εμπλέκονται σε διάφορα μονοπάτια σηματοδότησης, οι οποίοι θα μπορούσαν να χρησιμοποιηθούν σαν προβλεπτικοί βιοδείκτες αντίστασης. Επίσης, αυτοί οι βιοδείκτες θα μπορούσαν να χρησιμοποιηθούν στην επιλογή ασθενών που μπορεί να έχουν όφελος από επιπρόσθετες στοχεύουσες θεραπείες.
Η αντίσταση στην ταμοξιφαίνη είναι ο κύριος λόγος που περιορίζει την αποτελεσματικότητα της θεραπείας. Η θεραπεία με αναστολείς αρωματάσης (είτε ως αρχική θεραπεία είτε ως επακόλουθη της ταμοξιφαίνης) μπορεί να είναι δραστική σε ασθενείς που παρουσιάζουν αντίσταση στην ταμοξιφαίνη. Παρόλα αυτά, στον προχωρημένο καρκίνο μαστού, το ποσοστό ανταπόκρισης στους αναστολείς αρωματάσης είναι ελάχιστα μεγαλύτερο συγκρινόμενο με την ταμοξιφαίνη και ομοίως παρατηρείται πρωτογενής και επίκτητη αντίσταση. Τέλος, η φουλβεστράνη έχει επιδείξει κλινική δραστικότητα σε ασθενείς με καρκίνο μαστού που παρουσιάζουν δεύτερη υποτροπή μετά την αρχική ανταπόκριση στην ταμοξιφαίνη και στους αναστολείς αρωματάσης (1-3).
Παρά την έυρεση πιο ισχυρών αντιοιστρογονικών θεραπειών, η αντίσταση σε όλες τις μορφές ενδοκρινικής θεραπείας στον καρκίνο του μαστού αποτελεί μείζον πρόβλημα. Η κατανόηση των υπεύθυνων για την αντίσταση μοριακών μηχανισμών θα επιτρέψει τη χρήση νέων στρατηγικών μεθόδων θεραπευτικής αντιμετώπισης. Το κεφάλαιο αυτό παραθέτει αρχικώς μια περίληψη της λειτουργίας των οιστρογονικών υποδοχέων και ακολούθως επικεντρώνεται στην ανάλυση των μηχανισμών αντίστασης στην ορμονική θεραπεία στον καρκίνο μαστού και των πιθανών τρόπων αντιμετώπισής της.
Λειτουργία των οιστρογονικών υποδοχέων
Τα οιστρογόνα παίζουν σημαντικό ρόλο στην αύξηση, διαφοροποίηση και λειτουργία διαφόρων ιστών, συμπεριλαμβανομένων του μαζικού αδένα, του ενδομητρίου, των οστών, του καρδαγγειακού συστήματος και του εγκεφάλου. Ασκούν τη δράση τους μέσω δύο ενδοκυττάριων υποδοχέων, του ERα και του ERβ. Οι υποδοχείς ERα και ERβ κωδικοποιούνται από ξεχωριστά γονίδια που εντοπίζονται στα χρωμοσώματα 6 και 14 αντιστοίχως. Και οι δύο υποδοχείς ανευρίσκονται στο φυσιολογικό μαστό, αλλά μόνο ο υποδοχέας ERα συσχετίζεται με την ανάπτυξη καρκίνου του μαστού, ενώ ο ρόλος του ERβ δεν έχει διαλευκανθεί ακόμα. Παρόλα αυτά, μελέτες υποστηρίζουν ότι ο ERβ ασκεί αντίθετη δράση απ’ ότι ο ERα, αναστέλλοντας την ικανότητα των οιστρογόνων να διεγείρουν τον πολλαπλασιασμό των καρκινικών κυττάρων. Παρομοίως, έχει βρεθεί ότι η δυσλειτουργία του ERβ συντελεί στην καρκινογένεση και αντιστοίχως ότι η υπερέκφραση του συνδέεται με καλύτερη πρόγνωση.
Οι δύο υποδοχείς παρουσιάζουν κοινή αρχιτεκτονική δομή και αποτελούνται από έξι τομείς (Σχήμα 1). Το Ν-τελικό άκρο του τομέα Α/Β περιλαμβάνει μια περιοχή υπεύθυνη για τη μεταγραφική δραστηριότητα του ER σε απουσία του συνδέτη, η οποία ονομάζεται AF1. Ο τομέας C, που φέρεται με τον όρο DBD (DNA-binding domain), παίζει σημαντικό ρόλο στο διμερισμό του υποδοχέα και στη σύνδεση του με αλληλουχίες DNA. O τομέας D παίζει ρόλο στην εντόπιση του πυρήνα. Ο τομέας Ε, που αναφέρεται ως LBD (ligand binding domain),εμπεριέχει μια θέση διμερισμού του υποδοχέα και την περιοχή AF2, υπεύθυνη για την ενεργοποίηση του υποδοχέα που προκαλείται από το συνδέτη. Ο τομέας F είναι μια μικρή περιοχή που συντονίζει τις περιοχές AF1 και AF2, αν και δεν είναι απαραίτητος για την ενεργοποίηση της μεταγραφής. Οι δύο υποδοχείς ER παρουσιάζουν ομολογία μεταξύ των τομέων DBD και LBD.
O υποδοχέας ERα συσχετίζεται με τον πολλαπλασιασμό των κυττάρων και την επιβίωση μέσω δύο μηχανισμών, του επονομαζόμενου «κλασικού» και του «μη κλασικού». Ο κλασικός μηχανισμός αρχίζει με τη σύνδεση των οιστρογόνων στον υποδοχέα, η οποία οδηγεί στο διμερισμό του και στην έναρξη μεταγραφικής δραστηριότητας. Το σύμπλοκο οιστρογόνων–υποδοχέα συνδέεται άμεσα ή μέσω πρωτεϊνικών αλληλεπιδράσεων με μια αλληλουχία DNA γνωστή ως «στοιχείο απάντησης οιστρογόνων» (estrogen-response element-ERE), με αποτέλεσμα την επιστράτευση ρυθμιστικών πρωτεϊνών που ενισχύουν ή καταστέλλουν τη μεταγραφική δραστηριότητα του υποδοχέα, ανάλογα με την ειδικότητα του συνδέτη. Στο μη κλασικό μηχανισμό, η απάντηση στα οιστρογόνα διαμεσολαβείται μέσω υποδοχέων ER που εντοπίζονται στην πλασματική μεμβράνη ή σε άλλες περιοχές της μεμβράνης. Ο μεμβρανικός υποδοχέας ER αλληλεπιδρά με διάφορες πρωτεΐνες συμπεριλαμβανομένων G-πρωτεϊνών, υποδοχέων αυξητικών παραγόντων (EGFR, IGFR1, HER2) και κυτταροπλασματικών κινασών (MAPKs, P13K,AKT) και κινητοποιεί μηχανισμούς ανεξάρτητους από τη μεταγραφή γονιδίων. Πρόσφατα δεδομένα υποστηρίζουν ότι η πρωτεΐνη GPER μπορεί να παίζει ρόλο στη διαδικασία του μη κλασικού μηχανισμού απάντησης στα οιστρογόνα. Οι δύο μηχανισμοί απάντησης στα οιστρογόνα αλληλεπιδρούν μεταξύ τους και εμπέκονται στην ανάπτυξη και εξέλιξη του καρκίνου του μαστού (4-6).
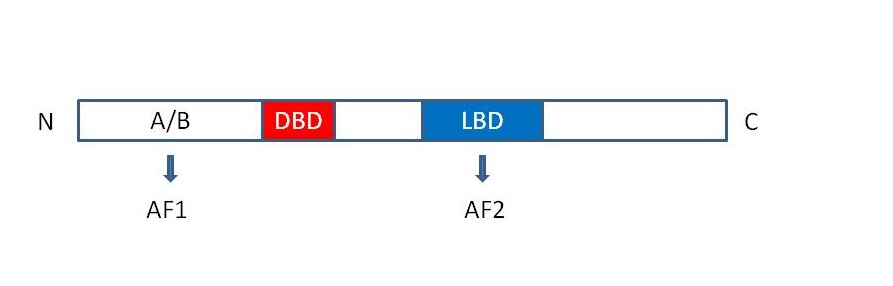
Σχήμα 1. Δομή οιστρογονικού υποδοχέα.
Ορμονική θεραπεία
Η ορμονική θεραπεία στον ορμονοεξαρτώμενο καρκίνο του μαστού περιλαμβάνει το χειρισμό του ενδοκρινικού συστήματος μέσω εξωγενούς χορήγησης φαρμάκων που εμποδίζουν την παραγωγή ή τη δραστηριότητα των οιστρογόνων.
Τα φάρμακα που ανήκουν στην κατηγορία SERMs και SERDs επηρεάζουν άμεσα τον υποδοχέα ER. Μετά τη σύνδεση ενός αναστολέα SERM στον υποδοχέα ER, ο μεταγραφικός παράγοντας μπορεί να συνδεθεί με το DNA, αλλά εμποδίζεται η έναρξη της μεταγραφής λόγω μιας αλλαγής στη δομή του υποδοχέα. H ταμοξιφαίνη, η οποία ανήκει σ αυτή την κατηγορία φαρμάκων, ανταγωνίζεται τη μεταγραφή που επάγεται από την περιοχή AF2, ενώ η μεταγραφή που διαμεσολαβείται από την περιοχή AF1 διατηρείται. Τα φάρμακα που ανήκουν στην κατηγορία SERDs, όπως η φουλβεστράνη, συνδέονται με την περιοχή LBD και αναστέλλουν το διμερισμό του υποδοχέα, με αποτέλεσμα να μειώνεται η ικανότητα του να συντονίζει τη μεταγραφή και να προωθείται η αποδόμηση του από το πρωτεόσωμα.
Οι αναστολείς αρωματάσης έχουν διαφορετικό μηχανισμό δράσης. Είναι γνωστό ότι πριν την εμμηνόπαυση, η οιστραδιόλη προέρχεται κυρίως από τον ωοθηκικό ιστό. Στις μετεμμηνοπαυσιακές γυναίκες, η πτώση της οιστραδιόλης αποδίδεται στη μείωση στης σύνθεσής της από τις ωοθήκες, και τα επίπεδα της παραμένουν στο ένα δέκατο των επιπέδων των προεμμηνοπαυσιακών γυναικών. Η βιοσύνθεση της οιστραδιόλης στις μετεμμηνοπαυσιακές γυναίκες γίνεται κυρίως στους περιφερικούς ιστούς, όπου το ένζυμο αρωματάση καταλύει τη μετατροπή ανδρογόνων σε οιστρογόνα και πιο συγκεκριμένα της ανδροστενδιόνης σε οιστρόνη και της τεστοστερόνης σε οιστραδιόλη. Στις μετεμμηνοπαυσιακές γυναίκες, τα οιστρογόνα που παράγονται σε περιφερικούς ιστούς αποτελούν σημαντικούς κινητοποιούς παράγοντες για τα καρκινικά κύτταρα του μαστού που εκφράζουν τον οιστρογονικό υποδοχέα. Οι στεροιειδικοί αναστολείς αρωματάσης, όπως η εξαμεστάνη, προσομοιάζουν και ανταγωνίζονται τα ενδογενή υποστρώματα. Συνδέονται μη αναστρέψιμα με την αρωματάση και την καθιστούν ανενεργή. Αντιθέτως, οι μη στεροειδικοί αναστολείς, δεσμεύονται αναστρέψιμα με την αρωματάση και εμποδίζουν την ενζυμική δραστηριότητα (7-10).
Ανταπόκριση στην ορμονική θεραπεία
Στον καρκίνο του μαστού, η ανοσοϊστοχημική ανίχνευση της έκφρασης του ER από τα καρκινικά κύτταρα αποτελεί το σημαντικότερο παράγοντα που καθορίζει την απάντηση στην ενδοκρινική θεραπεία, ενώ η έλλειψη έκφρασης ισοδυναμεί με αποτυχία της θεραπείας. Η πλειονότητα των καρκινικών κυττάρων που παρουσιάζουν εγγενή αντίσταση στην ορμονική θεραπεία δεν εκφράζουν ER. Παρόλα αυτά, πρωτογενής αντίσταση παρατηρείται και σε ένα μικρό ποσοστό των ER θετικών όγκων.
Πιο συγκεκριμένα, οι ER θετικοί όγκοι: α) δεν απαντούν πάντα στην ενδοκρινική θεραπεία β) παρουσιάζουν ετερογένεια απάντησης σε οποιοδήποτε ενδοκρινικό παράγοντα γ) μπορεί να εμφανίζουν ανθεκτικότητα σε μια κατηγορία ενδοκρινικής θεραπείας και ευαισθησία σε άλλες και δ) συχνά εξελίσσονται από ευαίσθητους σε ανθεκτικούς φαινοτύπους, παρόλο που διατηρούν την έκφραση του ER. Συνεπώς, όλες αυτές οι αντιφάσεις υποδεικνύουν ότι οι όγκοι που εκφράζουν ER αποτελούν μία ανομοιογενή υποομάδα με ποικίλλη ευαισθησία στην ορμονική θεραπεία. Επομένως, καθίσταται αναγκαία η εύρεση προβλεπτικών δεικτών για την αξιολόγηση του οφέλους της ορμονικής θεραπείας σε επιλεγμένους ασθενείς.
Ειναι γνωστό ότι ένα μέρος της ετερογένειας όσον αφορά στην ανταπόκριση στην ορμονική θεραπεία έχει αποδοθεί στo βαθμό της έκφρασης του ER, ο οποίος μπορεί να θεωρηθεί ποσοτικός προβλεπτικός παράγοντας απάντησης στην ορμονική θεραπεία. Υπάρχουν δεδομένα ότι τόσο η επικουρική θεραπεία με ταμοξιφαίνη όσο η ορμονική θεραπεία στη μεταστατική νόσο οδηγεί σε μεγαλύτερη βελτίωση των ποσοστών υποτροπής και θνησιμότητας σε όγκους με ισχυρότερη έκφραση ER. Σύμφωνα με οδηγίες του Διεθνούς Συνεδρίου Ειδικών για τον Καρκίνο Μαστού του St Gallen το 2009, όγκοι με ανοσοϊστοχημική έκφραση ER >50% θεωρούνται ότι παρουσιάζουν υψηλά ποσοστά ανταπόκρισης στην ορμονική θεραπεία. Από την άλλη μεριά, τα δεδομένα δύο κλινικών μελετών (ATAC και BIG 1-98) δείχνουν ότι δεν υπάρχει στατιστικώς σημαντική συσχέτιση μεταξύ των επιπέδων έκφρασης ER και της αποτελεσματικότητας των αναστολέων αρωματάσης σε σύγκριση με τη ταμοξιφαίνη. Συνεπώς, όσον αφορά στο όφελος απ’ την ορμονική θεραπεία, η θετικότητα των όγκων σε οιστρογονικούς υποδοχείς είναι πολύ πιο σημαντικός προβλεπτικός παράγοντας απ’ ότι ο βαθμός της θετικότητας. Παρόλα αυτά, είναι απαραίτητη η ανεύρεση αξιόπιστων προβλεπτικών βιοδεικτών πρωτογενούς αντίστασης στην ορμονική (de novo) θεραπεία στους ασθενείς με θετικούς ER όγκους.
Η ετερογένεια στην ανταπόκριση στην ορμονική θεραπεία μπορεί επίσης να οφείλεται ως ένα βαθμό στην αλληλεπίδραση του υποδοχέα ER με μονοπάτια σηματοδότησης αυξητικών παραγόντων και με διαφόρους ρυθμιστικούς παράγοντες. Για παράδειγμα, στους όγκους που εκφράζουν HER2, η ανταπόκριση στην ορμονική θεραπεία είναι μικρότερη, ακόμα και όταν υπάρχει υψηλή έκφραση ER. Παρόλα αυτά, διαφορές σε μοριακό επίπεδο έχουν διαφορετική επίδραση σε διαφορετικές κατηγορίες φαρμάκων. Πράγματι, σε κλινικές μελέτες αξιολόγησης της λετροζόλης, φάνηκε ότι η σχετική αποτελεσματικότητα της είναι μεγαλύτερη σε HER2 και EGFR θετικούς όγκους.
Συμπερασματικά, η ετερογένεια των ER θετικών όγκων ως προς την ανταπόκριση στη θεραπεία αποτελεί μια πρόκληση για έρευνα. Είναι απαραίτητη η εύρεση βιοδεικτών πρωτογενούς αντίστασης με σκοπό τη βελτίωση της επιλογής ασθενών και την εύρεση αυτών που δύνανται να επωφεληθούν εξαρχής από επιπρόσθετες θεραπείες (11-13).
Μηχανισμοί αντίστασης στην ορμονοθεραπεία και τρόποι αντιμετώπισης
Έχουν μελετηθεί διάφοροι μηχανισμοί αντίστασης στην ενδοκρινική θεραπεία και περιλαμβάνουν απώλεια ER υποδοχέα, σηματοδοτικά μονοπάτια, μεταβολή στην έκφραση των microRNAs καθώς και στο μεταβολισμό της ταμοξιφαίνης. Επιπλέον, τόσο η αλληλεπίδραση μεταξύ κλασικού και μη κλασικού μηχανισμού απάντησης στα οιστρογόνα, όσο και η αλληλοσυσχέτιση μεταξύ υποομάδων οιστρογονικών υποδοχέων και αυξητικών παραγόντων συντελούν στην αντίσταση στην ορμονοθεραπεία. Παρακάτω, περιγράφονται οι πιθανότεροι υπεύθυνοι μηχανισμοί.
1.Απώλεια ή τροποποίηση της έκφρασης του υποδοχέα ER
Όπως προαναφέρθηκε, η θετικότητα στους οιστρογονικούς υποδοχείς αποτελεί το κύριο προβλεπτικό παράγοντα ανταπόκρισης στην ορμονοθεραπεία, και η έλλειψη έκφρασης του ER αποτελεί τον κυρίαρχο μηχανισμό πρωτογενούς αντίστασης. Έχουν προταθεί διάφοροι μηχανισμοί που εξηγούν την απουσία της έκφρασης οιστρογονικών υποδοχέων. Επιγενετικές μεταβολές, όπως η μεθυλίωση των περιοχών CpG του υποκινητή του ΕR γονιδίου και η τροποποίηση των ιστονών, οδηγούν σε μια συμπαγή δομή του νουκλεοσώματος, η οποία περιορίζει τη μετάφραση. Σε in vitro πειράματα, έχει βρεθεί ότι ο συνδυασμός θεραπείας με αναστολείς της DNA μεθυλτρανσφεράσης-1,(DNMT-1, όπως η 5-αζα-2-δεοξυτιδίνη), ενζύμου που καταλύει τη μεθυλίωση και αναστολείς των δεακετυλασών των ιστονών, (HDAC, όπως η τριχοστατίνη Α) ενζύμων που αντιστρέφουν την ακετυλίωση των ιστονών, προκαλεί ενεργοποίηση της έκφρασης του ER στα καρκινικά κύτταρα του καρκίνου του μαστού και αποκαθιστά την ευαισθησία στα αντιοιστρογόνα. Επιλέον, σε In vitro και in vivo μελέτες, η θεραπεία με τον αναστολέα HDAC entinostat οδήγησε σε αύξηση της έκφρασης του ER και της αρωματάσης και επακολούθως σε ευαισθητοποίηση των καρκινικών κυττάρων στα οιστρογόνα και τη λετροζόλη. Επίσης, ένας άλλος αναστολέας HDAC, ο Scriptaid, έχει βρεθεί ότι προκαλεί την επανέκφραση ενός λειτουργικού υποδοχέα ER, εμποδίζει τον κυτταρικό πολλαπλασιασμό και επανευαισθητοποιεί τα ορμονοάντοχα καρκινικά κύτταρα στην ταμοξιφαίνη.
Από την άλλη μεριά, υποστηρίζεται ότι η απώλεια της έκφρασης ER μπορεί να ευθύνεται για την επίκτητη αντίσταση στην ορμονοθεραπεία. Παρόλα αυτά, μόνο 17-28% των ασθενών με επίκτητη αντίσταση στην ορμονοθεραπεία δεν παρουσιάζουν θετικότητα στον ER, και επιπλέον, οι ER (+) ασθενείς που αρχικά ανταποκρίνονται στην ταμοξιφαίνη, συνήθως δεν χάνουν την οιστρογονική έκφραση μετά την ανάπτυξη αντίστασης στη θεραπεία. Περίπου 20% των ασθενών απαντούν σε δεύτερης γραμμής θεραπεία με αναστολείς αρωματάσης ή φουλβεστράνη, το οποίο αποδεικνύει ότι εξακολουθούν να εκφράζουν οιστρογονικούς υποδοχείς.
Άλλοι μηχανισμοί που εμπλέκονται στην απώλεια της έκφρασης ER είναι η υποξία, η υπερέκφραση του EGFR ή του HER2, η ενεργοποίηση των MAPΚ κινασών, και τα γονίδια p53 και pRb2/p130. Η υποξία προκαλεί αποδόμηση του ER σε καρκινικά κύτταρα μέσω του πρωτεοσώματος. Σε ER αρνητικά κύτταρα, έχει βρεθεί υψηλή έκφραση EGFR και HER2, υπονοώντας ότι η ενεργοποίηση της σηματοδότησης αυξητικών παραγόντων και ακολούθως, της MAPK κινάσης μπορεί να συντελεί στη μεταγραφική καταστολή του γονιδίου ER και συνεπώς, στην αντίσταση στην ενδοκρινική θεραπεία. Από την άλλη μεριά, πρόσφατες έρευνες έχουν δείξει ότι μεταλλάξεις στο p53 απορρυθμίζουν την έκφραση του γονιδίου ER. Άλλωστε, ένα μεγάλο ποσοστό των όγκων του μαστού με μεταλλάξεις στο p53 είναι ER αρνητικοί. Επίσης,το γονίδιο pRb2/p130 παίζει σημαντικό ρόλο στη ρύθμιση της μεταγραφής του υποκινητή του ER γονιδίου, καθιστώντας το υποσχόμενο μόριο στόχευσης στη θεραπεία του καρκίνου του μαστού, ειδικά σε ER αρνητικούς όγκους.
Ένας άλλος μηχανισμός που τροποποιεί την έκφραση ER είναι οι μεταλλάξεις γονιδίων που επιδρούν στην ανταπόκριση στα αντιοιστρογόνα. Για παράδειγμα, η αντικατάσταση της ασπαρτάσης από την τυροσίνη στη θέση 537 όπου δεσμεύεται ο συνδέτης, προκαλεί ενεργοποίηση του υποδοχέα ER απουσία του συνδέτη. Επίσης, τροποποίηση της λυσίνης 303 σε αργινίνη οδηγεί σε αυξημένη ευαισθησία στα οιστρογόνα, αλλά πιθανόν να μεταβάλλει τη λειτουργία ER. Παρόλα αυτά, οι μεταλλάξεις αυτές απαντώνται σε 1% των καρκίνων μαστού.
Τέλος, αντίσταση στην ταμοξιφαίνη έχει παρατηρηθεί σε τροποποιημένη έκφραση του υποδοχέα ERβ.Υψηλά επίπεδα ERβ έχουν βρεθεί σε προδιηθητικούς καρκίνους του μαστού ασθενών με αντίσταση στην ταμοξιφαίνη.Παρόλα αυτά, άλλες μελέτες έχουν δείξει ότι χαμηλά επίπεδα του ERβ συντελούν στην αντίσταση στην ορμονοθεραπεία. Συνεπώς, ο ρόλος του ERβ στην αντίσταση στην ορμονοθεραπεία παραμένει αμφιλεγόμενος (14-17).
2.Επιγενετικοί μηχανισμοί που ρυθμίζουν τη έκφραση ER
Διάφοροι επιγενετικοί μηχανισμοί που ρυθμίζουν την έκφραση διαφόρων γονιδίων ρυθμίζουν και την έκφραση του ER. Μελέτες που επικεντρώνονται στην επιγενετική μπορούν να οδηγήσουν στην ανακάλυψη βιοδεικτών που προβλέπουν και διαγιγνώσκουν την επίκτητη αντίσταση στην ορμονοθεραπεία.
Η απώλεια της έκφρασης του ΕRα συχνά συσχετίζεται με την υπερμεθυλίωση του υποκινητή του γονιδίου ERα. Παρομοίως, στους όγκους που παρουσιάζουν αντίσταση στην ορμονική θεραπεία παρατηρείται και μεθυλίωση του PR. Επίσης, η έκφραση της πρωτεΐνης HOXB13, η οποία έχει βρεθεί ότι σχετίζεται με υποτροπή σε ασθενείς με ER θετικούς όγκους και αρνητικούς λεμφαδένες που λαμβάνουν ταμοξιφαίνη, βρίσκεται υπό επιγενετικό έλεγχο. Από την άλλη μεριά, η μεθυλίωση του υποκινητή της κινάσης CDK10 (cyclin-dependent kinase 10) προκαλεί μείωση των επιπέδων της CDK10. Έχει βρεθεί ότι ασθενείς με χαμηλά επίπεδα CDK10 υποτροπιάζουν γρήγορα μετά από θεραπεία με ταμοξιφαίνη. Η μεθυλίωση του DNA υποψήφιων γονιδίων και η συσχέτιση τους με την πρόγνωση ασθενών με καρκίνο του μαστού που λαμβάνουν ταμοξιφαίνη αποτελεί αντικείμενο εντατικής έρευνας. Επίσης, αναστολείς ενζύμων που ελέγχουν επιγενετικές μεταβολές, όπως η DMNT και οι HDACs που προανεφέρθησαν, έχουν δείξει υποσχόμενη αντικαρκινική δράση. Δεδομένου ότι οι επιγενετικές μεταβολές είναι αναστρέψιμες, η αναστολή των μηχανισμών αυτών αποτελεί θεραπευτική στρατηγική για τη θεραπεία όγκων με αντίσταση στην ορμονική θεραπεία, όχι μόνο δρώντας άμεσα στις επιγενετικές αλλαγές, αλλά και συντονίζοντας γνωστούς στόχους άλλων θεραπειών (18-19).
3. Ρύθμιση σηματοδοτικών μονοπατιών
Τα σηματοδοτικά μονοπάτια των υποδοχέων των αυξητικών παραγόντων μπορούν να επάγουν την ανάπτυξη καρκίνου, είτε σε συντονισμό με το ER μονοπάτι είτε παρακάμπτοντας το, και υπάρχουν ενδείξεις ότι συσχετίζονται και με την αντίσταση στην ορμονοθεραπεία. Η ενίσχυση της μετάδοσης σημάτων μέσω των μονοπατιών αυτών ενεργοποιεί τόσο τον κλασικό όσο και το μη κλασικό μηχανισμό οιστρογονικής δραστηριότητας, ενώ τα περισσότερα αντι-οιστρογονικά φάρμακα ενεργοποιούν μόνο τον κλασικό μηχανισμό. Μεταξύ των μονοπατιών αυτών και του μονοπατιού σηματοδότησης του ER, υπάρχει αλληλεπίδραση, έτσι ώστε ο μεμβρανικός υποδοχέας ER να ενεργοποιεί τη σηματοδότηση μέσω αυξητικών παραγόντων και από την άλλη , η ενεργοποίηση αυτή να προωθεί τη φωφορυλίωση του ER και ρυθμιστικών πρωτεϊνών. Η φωσφωρυλίωση των σερινών 118 και 167 στην περιοχή AF1 του υποδοχέα ER προωθεί την επανενεργοποίηση της λειτουργίας του ER ανεξάρτητα από τη δέσμευση του συνδέτη και συντελεί στη μεταγραφή γονιδίων ευαίσθητων στα οιστρογόνα παρουσία αντιοιστρογονικών παραγόντων. Επιπλέον, έχει βρεθεί ότι τα σηματοδοτικά μονοπάτια των αυξητικών παραγόντων προωθούν τη φωσφορυλίωση του υποδοχέα ER και των συνενεργοποιητών του. Πράγματι, η υπερέκφραση του μορίου AIB1 έχει συσχετισθεί με αντίσταση στην ταμοξιφαίνη σε ασθενείς με καρκίνο μαστού και πιθανόν να αποτελεί υπεύθυνο μηχανισμό για την αντίσταση στην ταμοξιφαίνη σε κύτταρα MCF-7 που υπερεκφράζουν HER2.
Άλλωστε, στους όγκους με αντίσταση στην ορμονοθεραπεία, η υπερέκφραση και ενεργοποίηση υποδοχέων αυξητικών παραγόντων, όπως ο EGFR, ο ΗΕR2 και ο IGF1R, οδηγεί σε ενεργοποίηση των MAPK και PI3K/AKT σηματοδοτικών μονοπατιών και σε επακόλουθη άυξηση του κυτταρικού πολλαπλασιασμού. Πειραματικά δεδομένα έχουν δείξει ότι η γενετική επιμόλυνση ER θετικών καρκινικών κυττάρων μαστού με HER2 έχει ως αποτέλεσμα την μείωση των επιπέδων ER και την αντίσταση στην ταμοξιφαίνη. Επιπλέον, διάφορες μελέτες υποστηρίζουν ότι η έκφραση HER2 παίζει σημαντικό ρόλο στην αντίσταση στην ορμονοθεραπεία. Σε μια μεταανάλυση κλινικών μελετών, φάνηκε ότι οι ασθενείς με μεταστατικό ER/HER2(+) καρκίνο μαστού, ελάμβαναν θεραπεία με ταμοξιφαίνη συντομότερα μετά την αποτυχία της θεραπείας σε σχέση με τους HER2(-) όγκους. Ακόμη, σε όγκους που αποκτούσαν αντίσταση στη φουλβεστράνη παρατηρούνταν υπερέκφραση των μορίων HER2 και ΜΑPK, υποστηρίζοντας αυτό το μηχανισμό ως υπεύθυνο για την αντίσταση. Όσον αφορά την αντίσταση στην εξαμεστάνη, ένας προτεινόμενος μηχανισμός είναι η ενεργοποίηση του μονοπατιού EGFR μέσω της πρωτεΐνης αμφιρεγκουλίνης, με επακόλουθα την ενεργοποίηση του MAPK και την επαγωγή του κυτταρικού πολλαπλασιασμού.
Όπως προαναφέρθηκε, το μονοπάτι IGF1R αλληλεπιδρά στενά με το μονοπάτι ER. Πιο συγκεκριμένα, ο IGF1R προωθεί την ενεργοποίηση κινασών που φωσφορυλιώνουν τον ER. Eναλλάξ, τα οιστρογόνα ενισχύουν τη δράση του αυξητικού παράγοντα IGF2 που ενδυναμώνει τη σηματοδότηση μέσω του IGF1R. Έχει αποδειχθεί ότι ER (+) MC7 κύτταρα με έκτοπη έκφραση του υποδοχέα IGF1R παρουσιάζουν αντίσταση στη θεραπεία με ταμοξιφαίνη και τη φουλβεστράνη, αλλά μετά τη δέσμευση του συνδέτη IGF1 παρατηρείται ενεργοποίηση των μονοπατιών MAPK και PI3K ανεξαρτήτως σηματοδότησης του ER. Επιπλέον, οι όγκοι που παρουσιάζουν υψηλή έκφραση IGF1 και ER αναπτύσσουν αργότερα αντίσταση στην ταμοξιφαίνη, γεγονός που υποδεικνύει μια συνεργεια μεταξύ των δύο.
Τέλος, πρόσφατες ενδείξεις υποστηρίζουν το μόριο MED1, που αποτελεί συνενεργοποιητή του ER, μπορεί να ενοχοποιείται για την αντίσταση στην ταμοξιφαίνη σε HER2(+) ER(+) όγκους. Πιο συγκεκριμένα, μείωση των επιπέδων του MED1 οδηγεί σε ευασθητοποίηση των HER2(+) καρκινικών κυττάρων στη θεραπεία με ταμοξιφαίνη (20-25).
4. Μονοπάτι PI3K
To μονοπάτι PI3K έχει μελετηθεί διεξοδικά για το ρόλο του στην ογκογένεση. Το μόριο ΑΚΤ αποτελεί ένα μόριο στόχευσης του μονοπατιού που προωθεί τον κυτταρικό πολλαπλασιασμό. Έχει βρεθεί ότι η καταστολή του PTEN (αναστολέα του ΑΚΤ ) οδηγεί σε αυξημένη έκφραση φωσφορυλίωσης του ΑΚΤ σε ER(+) καρκινικά κύτταρα μαστού, με αποτέλεσμα την επαγωγή του κυτταρικού πολλαπλασιασμού και την αντίσταση στην ταμοξιφαίνη και τη φουλβεστράνη. Επιπλέον, κύτταρα που παρουσιάζουν αντίσταση στην ορμονοθεραπεία εμφανίζουν αυξημένη έκφραση επιπέδων IGF-1R, HER2 και EGFR, καθώς και αυξημένη ενεργοποίηση PI3K/AKT/mTOR. Αξίζει να σημειωθεί ότι παρόλο που η ενεργοποίηση του PI3K μέσω υπερέκφρασης HER2 ή FGFR2 επιφέρει αντίσταση στην ορμονοθεραπεία σε ΕR (+) όγκους, η παρουσία μεταλλάξεων στην καταλυτική υποομάδα του PI3K (PI3KCA), που ανευρίσκονται σε ποσοστό 30% των ορμονοευαίσθητων όγκων, συνάδει με καλύτερη πρόγνωση των όγκων και ο ρόλος τους δεν έχει επαρκώς διευκρινισθεί.
Μελέτες σε καρκινικά κύτταρα μαστού έχουν αποδείξει ότι η υποβολή σε μακροχρόνια στέρηση οιστρογόνων οδηγεί αρχικώς σε υπερευαισθησία σε χαμηλές δόσεις οιστρογόνων και ακολούθως τα κύτταρα καθίστανται μη εξαρτώμενα από τα οιστρογόνα. Αυτό το φαινόμενο συντελεί στην αντίσταση στην ορμονοθεραπεία, και πιθανώς διαμεσολαβείται από την αυξημένη έκφραση MAPK και mTOR. Παράγματι, κύτταρα MCF-7 που εκτίθενται σε μακροχρόνια χορήγηση ταμοξιφαίνης παρουσιάζουν αυξημένα επίπεδα MAPK κατά τη διάρκεια της απόκτησης αντίστασης στην ταμοξιφαίνη. Τα κύτταρα αυτά περνούν από τρία στάδια: α) η ταμοξιφαίνη συμπεριφέρεται ως ανταγωνιστής οιστρογόνων β) τα κύτταρα σταδιακά γίονται ευαίσθητα στις αγωνιστικές επιδράσεις της ταμοξιφαίνης και γ) τα κύτταρα καθιστώνται υπερευαισθητα στην ταμοξιφαίνη (26-28).
5. Μονοπάτι Stress-Activated Protein Kinase/ c-junNH2 kinase
To μονοπάτι Stress-Activated Protein Kinase/ c-junNH2 kinase αλληλεπιδρά με τον ER μέσω σύνδεσης με το σύμπλοκο μεταγραφής AP-1 ή μέσω άμεσης ενεργοποίησης του MAPK. Σε μια μελέτη, η ανάπτυξη αντίστασης στην ταμοξιφαίνη συσχετίσθηκε με αυξημένη σύνδεση του AP-1 στο DNA, ενώ άλλα δεδομένα σε xenografts υποστηρίζουν ότι οι όγκοι που παρουσιάζουν αντίσταση στην ταμοξιφαίνη εμφανίζουν αυξημένη μεταγραφική δραστηριότητα του AP-1.
Eπιπλέον, έχει βρεθεί ότι καρκινικά κύτταρα με αντίσταση στην ταμοξιφαίνη, χαρακτηρίζονται από υψηλά επίπεδα p38 MAPK. H πρωτεΐνη αυτή ενεργοποιείται σε απάντηση στην ιονίζουσα ακτινοβολία, το οξειδωτικό στρες και την ισχαιμία των ιστών. Δεδομένου ότι σε κύτταρα ενδομητρίου η φωσφορυλίωση του p38 αυξάνει την αγωνιστική δράση της ταμοξιφαίνης, τα αυξημένα επίπεδα p38 MAPK μπορεί να αποτελούν τον υπεύθυνο μηχανισμό για την αγωνιστική δράση της ταμοξιφαίνης (29-31).
6. Άλλα σηματοδοτικά μονοπάτια
Σ’ αυτά συμπεριλαμβάνονται ο παράγοντας nuclear factor-κΒ (NFκΒ), ο Notch, ο αυξητικός παράγοντας της κερατίνης (keratinocyte growth factor-KGF) και ο αυξητικός παράγοντας των αιμοπεταλίων (PDGF).
O ΝFκΒ προωθεί τον κυτταρικό πολλαπλασιασμό και την ενεργοποίηση πρωτεϊνών όπως η κυκλίνη D1 και ο ενεργοποιητής του πλασμινογόνου (uPA). Η ενεργοποίηση του έχει συσχετισθεί με πρόοδο νόσου στους μη ομονοεξαρτώμενους όγκους. Στους ΕR(+) όγκους, οι οποίοι παρουσιάζουν χαμηλά επίπεδα NFκΒ, έχει βρεθεί ότι ο NFκB ενεργοποιείται από την ταμοξιφαίνη και επάγει τον κυτταρικό πολλαπλασιασμό, συντελώντας στην αντίσταση στην ορμονοθεραπεία.
Το μονοπάτι KGF προκαλεί ανάπτυξη του επιθηλίου του μαστού επάγοντας τη δραστηριότητα της αρωματάσης, προωθώντας έτσι τη μετατροπή ανδρογόνων σε οιστρογόνα σε ανθρώπινα κύτταρα μαστού. Επίσης, προκαλεί μείωση των επιπέδων ΕR και PR συντελώντας έτσι στην αντίσταση στην ορμονοθεραπεία. Από την άλλη μεριά, το μονοπάτι Notch εμπλέκεται στη ρύθμιση αρχέγονων καρκινικών κυττάρων (Cancer Stem Cells-CSCs) στο καρκίνο του μαστού. Πιο συγκεκριμένα, ο παράγοντας Notch συνδέεται με τον αντίστοιχο υποδοχέα, ,μεταφέρεται στον πυρήνα όπου συνδέεται με άλλα πρωτεϊνικά μόρια και επάγει τη μεταγραφή γονιδίων, ρυθμίζοντας έτσι τη μετανάστευση και επέκταση των καρκινικών κυττάρων. Η οιστραδιόλη εμποδίζει τη δραστηριότητα του. Αντιθέτως, η ταμοξιφαίνη επανενεργοποιεί τον Notch. H φαρμακευτική αναστολή του Notch (μέσω αναστολέων ειδικών ενζύμων, των γ-σικρετασών) είναι πιο αποτελεσματική σε συνδυασμό με τη ταμοξιφαίνη και φαίνεται ότι μπορεί να μπλοκάρει την αναπαραγωγική δράση της ταμοξιφαίνης μέσω του Notch, επιτρέποντάς την να ασκήσει την ανταγωνιστική της δράση και συντελώντας στην καταπολέμηση της αντίστασης.
Όσον αφορά στον PDGFR, αποτελεί έναν υποδοχέα τυροσινικής κινάσης που ενεργοποιείται μετά τη σύνδεση του PDGF και παίζει σημαντικό ρόλο στην απαγωγή διαφόρων ενδοκυττάριων μονοπατιών και στον κυτταρικό πολλασπλασιασμό. Η Abl είναι μια πρωτεϊνική κινάση του μονοπατιού που εμπλέκεται τη ρύθμιση της απόπτωσης και της απάντησης στο στρες. Μελέτες in vitro και in vivo έχουν αποδείξει ότι η αλληλεπίδραση ER και PDGF/Abl μονοπατιού μπορεί να παίζει ρόλο στη δημιουργία ανθεκτικού στην ορμονοθεραπεία φαινοτύπου (32-33).
7. Τροποποιημένη έκφραση συγκεκριμένων microRNAs
Tα microRNAs αποτελούν μικρά ρυθμιστικά μόρια RNA που περιλαμβάνουν 19-24 νουκλεοτίδια και ρυθμίζουν την έκφραση συγκεκριμένων πρωτεϊνών με βάση συμπληρωματικές αλληλουχίες με τα προς στόχευση μόρια mRNA. Aυτή η διαδικασία επιτυγχάνεται είτε με αποδόμηση του μορίου mRNA είτε με καταστολή της πρωτεϊνικής σύνθεσης. Μ’ αυτόν τον τρόπο, τα microRNAs ρυθμίζουν μια ποικιλλία κυτταρικών διαδικασιών όπως ο κυτταρικός πολαπλασιασμός και ο κυτταρικός θάνατος και εμπλέκονται στην καρκινογένεση. Τροποποιημένη έκφραση τους έχει ενοχοποιηθεί στην ανάπτυξη αντίστασης στην ταμοξιφαίνη, καθώς διαφορετική έκφραση microRNAs (miRs) έχει ταυτοποιηθεί σε κυτταρικές σειρές καρκίνου μαστού που παρουσιάζουν αντίσταση στην ταμοξιφαίνη απ΄ότι αυτές που εμφανίζουν ευαισθησία. Για παράδειγμα, τα επίπεδα miR221/222 παρατηρούνται αυξημένα στους HER2(+) όγκους και οδηγούν σε αντίσταση στη ταμοξιφαίνη σε ορμονοευαίσθητα κύτταρα καρκίνου του μαστού, μέσω μείωσης των επιπέδων ενός αναστολέα κυτταρικού κύκλου. Επίσης, διαταραγμένη έκφρασή του έχει συσχετισθεί με αντίσταση στη φουλβεστράνη, μέσω ενεργοποίησης της β-κατενίνης. Αλλα μόρια microRNAs που ενοχοποιούνται στην αντίσταση στην ορμονοθεραπεία αποτελούν τα miR-342, miR-451 και miR-210. Τέλος, το miR-128a, που στοχεύει τον υποδοχέα TGFβ, έχει συσχετισθεί με αντίσταση στη λετροζόλη (34-35).
8.Ρυθμιστικές πρωτεΐνες
Οι ρυθμιστικές πρωτεΐνες επηρεάζουν τη ρύθμιση της μεταγραφικής δραστηριότητας του ER, επιτρέποντας τη δράση του ως αγωνιστή σε φυσιολογικούς ιστούς, αλλά ως ανταγωνιστή στον καρκίνο μαστού. Όμως, υψηλά επίπεδα συνενεργοποιητών του ER, όπως ο ΑΙΒ1, ενισχύουν τη δραστηριότητα της ταμοξιφαίνης ως αγωνιστή και μπορεί να συντελούν στην αντίσταση στην ταμοξιφαίνη. Αντιστοίχως, η απομάκρυνση του AIB από τον ER έχει αποδειχθεί ότι αποκαθιστά την ευαισθησία των κυττάρων στην ταμοξιφαίνη. Σε άλλες μελέτες, ο AIB1 έχει συσχετισθεί με αντίσταση στην ταμοξιφαίνη σε HER2(+) όγκους. Ένα άλλο μόριο , ο παράγοντας Pax2, αποτελεί σημαντικό διαμεσολαβητή της καταστολής του HER2 σε ER(+) όγκους, και θεωρείται ότι η αναστολή του Pax2 οδηγεί σε αυξημένα επίπεδα HER2 και εμποδίζει τη δράση της ταμοξιφαίνης. Τέλος, ο παράγοντας nuclear receptor corepressor (NCoR) αποτελεί σημαντικό προβλεπτικό παράγοντα ανταπόκρισης στην ταμοξιφαίνη. Έχει βρεθεί ότι μειωμένα επίπεδα του σχετίζονται με επίκτητη αντίσταση στην ταμοξιφαίνη και ότι ορμονοευαίσθητα κύτταρα εμφανίζουν υψηλό ποσοστό δέσμευσης του NCoR στον ER (36-37) .
9. Μεταβολισμός της ταμοξιφαίνης και αντίσταση με γενετικό υπόβαθρο
Είναι γνωστό ότι η ταμοξιφαίνη μεταβολίζεται στο ήπαρ από τα κυτοχρώματα CYP3A4 και CYP2D6, τα οποία παράγουν Ν-δεσμεθυλταμοξιφαίνη και 4-υδροξυταμοξιφαίνη αντίστοιχα. Στη συνέχεια, περαιτέρω οξείδωση των μεταβολιτών έχει ως αποτέλεσμα την παραγωγή ενός ενεργού μεταβολίτη, γνωστού ως ενδοξιφαίνη, που καταλύεται απο το CYP2D6. H ενδοξιφαίνη καταστέλλει τον κυτταρικό πολλαπλασιασμό πολύ περισσότερο από την ταμοξιφαίνη και θεωρείται ο υπεύθυνος μεταβολίτης για την αντιοιστρογονική της δραστηριότητα. Συνεπώς, πολυμορφισμοί στο κυτόχρωμα CYP2D6 μπορεί να επηρεάσουν την ενζυμική δραστηριότητα του. Κατ’αυτόν τον τρόπο, παράγονται διαφορετικοί μεταβολίτες της ταμοξιφαίνης, με αποτέλεσμα διαφορετικές θεραπευτικές ανταποκρίσεις στις ίδιες δόσεις φαρμάκου. Η γενετική ταυτοποίηση του CYP2D6 θα βοηθούσε στη σωστή επιλογή και δοσολογία της ταμοξιφαίνης.
Επιπλέον, παρόλο που η ταμοξιφαίνη μεταβολίζεται στο ήπαρ, καρκινiκά κύτταρα μπορεί να εμπλέκονται σ’αυτή τη διαδικασία, εκφράζοντας λειτουργικό κυτόχρωμα P450. Έτσι, η διαταραγμένη ικανότητα ενός όγκου να μεταβολίζει την ταμοξιφαίνη μπορεί να συντελεί στη αντίσταση. Προς αντιμετώπιση αυτού του μηχανισμού αντίστασης, έχει δοκιμαστεί η εκλεκτική αύξηση της έκθεσης των καρκινικών κυττάρων σε ενεργούς μεταβολίτες που παράγονται από ένα εξωγενές P450 γονίδιο (γενετική θεραπεία). Τα πλεονεκτήματα αυτής της θεραπευτικής στρατηγικής είναι η ενισχυμένη θεραπευτική αποτελεσματικότητα χωρίς αύξηση της τοξικότητας του ξενιστή και η πιθανότητα χορήγησης αποτελεσματικών αντικαρκινικών προφαρμάκων (38-39).
Αξίζει να σημειωθεί ότι η συγχορήγηση ταμοξιφαίνης με φάρμακα που αναστέλλουν τη δράση του κυτοχρώματος CYP2D6, όπως ορισμένα αντικαταθλιπτικά, μειώνουν την παραγωγή ενδοξιφαίνης και ακολούθως τη δραστικότητα της ταμοξιφαίνης.
Οι μηχανισμοί αντίστασης στην ορμονοθεραπεία και οι αντίστοιχοι τρόποι αντιμετώπισής τους παρουσιάζονται στον πίνακα 2.
| Παράγοντες που επηρεάζουν | Μηχανισμός | Παραδείγματα | Τρόπος αντιμετώπισης |
| Αυξητικοί παράγοντες και κινάσες | Επηρεάζουν τις μεταβολές του ER και των ρυθμιστικών πρωτεϊνών μετά τη μετάφραση, ενεργοποιούν τον «μη κλασικό μηχανισμό» οιστρογονικής δραστηριότητας | EGFR/HER2,IGFR-1, MAPK, PI3K/ΑΚΤ/mTOR PDGF, KGF,CDK4/6, Νotch | Gefitinib, lapatinib, Pan/PI3K και AKT αναστολείς, everolimus, palbociclib, αναστολείς γ-σικρετασών |
| Επιγενετική | Μεθυλίωση υποκινητών, αποακετυλίωση ιστονών, απώλεια έκφρασης ERa | Ιστόνες, HOXB13, CDK10 | Aναστολείς HDAC (erinostat) και DMNT |
| Μεταγραφικοί παράγοντες | Προκαλούν μη εξαρτώμενη από οιστρογόνα ανάπτυξη του όγκου | ΕRβ, NFκΒ, ΑP-1 | Αναστολείς NFκB |
| Ρυθμιστικές πρωτεΐνες | Kαθορίζουν αν οι εκλεκτικοί τροποποιητές των ER δρουν ως αγωνιστές ή ως ανταγωνιστές | ΑΙΒ1, ΝCOR | |
| microRNAs | Tροποποιημένη έκφραση | miR221/222, miR-342, miR-451, miR-210, miR-128a | |
| Γενετικές ανωμαλίες | Μεταλλάξεις στο κυτόχρωμα CYP450 εμποδίζουν τη μετατροπή της ταμοξιφαίνης στον ενεργό μεταβολίτη της ενδοξιφαίνη | CYP3A4, CYP2D6 | Γενετική θεραπεία |
Πίνακας 2. Μηχανισμοί αντίστασης στην ορμονοθεραπεία.
AIB1: amplified in breast 1, AP1: activator protein 1, CYP450: κυτόχρωμα p450, EGFR: epidermal growth factor receptor, ER:estrogen receptor (οιστρογονικός υποδοχέας), IGF1-R: insulin-like growth factor-1 receptor, MAPK: mitogen-activated protein kinase, NCOR1: co-repressor of estrogen receptor, PI3K: phosphatidylinositol-3 kinase.
Τρόποι αντιμετώπισης της αντίστασης
Η εμφάνιση αντίστασης στην ορμονοθεραπεία οδηγεί στην ανάπτυξη θεραπευτικών στρατηγικών, που α) ανταγωνίζονται τον ER, ενώ δεν επικεντρώνονται στην κατάργησης της εξαρτώμενης από τα οιστρογόνα δραστηριότητας και β) στοχεύουν μονοπάτια αυξητικών παραγόντων και την αλληλεπίδραση τους με τον ER στην δημιουργία αντίστασης στην ορμονοθεραπεία.
1.Συνδυασμός θεραπείας με φουλβεστράνη
Η θεραπεία με αναστολείς αρωματάσης δεν εμποδίζει πλήρως τη σηματοδότηση που διαμεσολαβείται από τα οιστρογόνα στον όγκο και για το λόγο αυτό ο όγκος μπορεί να συνεχίζει να εκφράζει ER και PR. Η μεγιστοποίηση του αποτελέσματος της αντιοιστρογονικής θεραπείας μπορεί να επιτευχθεί με θεραπευτικούς συνδυασμούς που στοχεύουν τον μη συνδεδεμένο ER υποδοχέα (μη κλασικός μηχανισμός). Οι διάφορες κατηγορίες ενδοκρινικών θεραπειών παρουσιάζουν ατελώς αλληλοεπικαλυπτόμενους μηχανισμούς δράσης, μεγαλώνοντας την πιθανότητα βελτιωμένης αποτελεσματικότητας με το συνδυασμό των θεραπειών. Για παράδειγμα, ο συνδυασμός με φουλβεστράνη μπορεί να συντελέσει σε πλήρη αποκλεισμό της σηματοδότησης από οιστρογόνα. Παρόλα αυτά, σε μια μεγάλη τυχαιοποιημένη μελέτη σε μη θεραπευθέντες ασθενείς με ER (+) όγκους, η οποία συνέκρινε τη χορήγηση αναστραζόλης, φουλβεστράνης ή του συνδυασμού τους, δεν παρατηρήθηκαν διαφορές σε σχέση με την έκφραση PR, ΕR και Ki67.
Δεν έχει αποσαφηνιστεί ποια είναι η κατάλληλη θεραπεία μετά από αποτυχία ενός μη στεροειδικού αναστολέα αρωματάσης. Στην τυχαιοποιημένη μελέτη EFECT (Εvaluation of Faslodex and Examestane Clinical Trial), η φουλβεστράνη και η εξαμεστάνη είχαν παρόμοια αποτελέσματα. Άλλωστε, η τυχαιοποιημένη φάσης ΙΙΙ μελέτη SOFEA (Study of Faslodex with or without concomitant Arimidex vs. Examestane following progression on non-steroidal Aromatase inhibitors), διενεργήθηκε σε μετεμμηνοπαυσιακές γυναίκες μετά από υποτροπή σε μη στεροειδικό αναστολέα αρωματάσης. Στη μελέτη αυτή , δεν παρατηρήθηκε διαφορά στη ολική επιβίωση ή την επιβίωση ελεύθερης νόσου μεταξύ εξαμεστάνης, φουλβεστράνης, ή στον συνδυασμό αναστραζόλης και φουλβεστράνης. Τέλος, στη μελέτη FACT (Fulvestrant and Anastrazole Combination therapy), γυναίκες με ER θετικούς όγκους τυχαιοποιήθηκαν να λάβουν αναστραζόλη ή το συνδυασμό αναστραζόλης και φουλβεστράνης κατά την πρώτη υποτροπή,. Ο συνδυασμός των δύο ορμονοθεραπειών έδωσε καλύτερα αποτελέσματα.
Ο συνδυασμός με φουλβεστράνη, έχει δοκιμαστεί και στο μεταστατικό καρκίνο μαστού. Σε μια τυχαιοποιημένη μελέτη, μετεμμηνοπαυσιακές γυναίκες που δεν είχαν λάβει καμία προηγούμενη θεραπεία, είχαν όφελος στη συνολική επιβίωση με το συνδυασμό φουλβεστράνης και αναστραζόλης. Τα δεδομένα αυτά υπογραμμίζουν τη σημαντικότητα της στόχευσης της δραστηριότητας του ΕR με τον μη κλασικό μηχανισμό, δηλαδή ανεξάρτητα από τη δέσμευση του συνδέτη (40-43).
2. Συνδυασμός θεραπείας με αναστολείς σηματοδοτικών μονοπατιών αυξητικών παραγόντων
Όπως προαναφέρθηκε, η σηματοδότηση μέσω αυξητικών παραγόντων επηρεάζει τη δραστικότητα του ER. Ο στόχος του συνδυασμού της ορμονοθεραπείας με αναστολείς σηματοδοτικών μονοπατιών αυξητικών παραγόντων είναι η αύξηση της ανταπόκρισης στην ορμονοθεραπεία, η καθυστέρηση ή ακόμα και η αποτροπή της εμφάνισης αντίστασης και σε περιπτώσεις εγκατεστημένης αντίστασης, η αποκατάσταση της ορμονικής ευαισθησίας. Αρχικώς, πειραματικά δεδομένα με αναστολείς τυροσινικής κινάσης, ανέδειξαν υποσχόμενα αποτελέσματα. Ακολούθησαν φάσης ΙΙ μελέτες, όπως για παράδειγμα μια μελέτη από τον Smith και τους συνεργάτες του, η οποία απέδειξε ότι στον ορμονοεξαρτώμενο καρκίνο μαστού, ο προεγχειρητικός συνδυασμός του EFGR αναστολέα gefitinib με αναστραζόλη δεν υπερνικά την αντίσταση. Αντιθέτως, στο μεταστατικό HER2/ER θετικό καρκίνο μαστού, ο συνδυασμός lapatinib, ενός αναστολέα τυροσινικής κινάσης που μπλοκάρει τον EGFR, με λετροζόλη, έδειξε κλινικό όφελος. Αξίζει να σημειωθεί ότι οι ασθενείς με HER2(-)/ ER(+) όγκους δεν επωφελήθηκαν από το συνδυασμό αυτό της θεραπείας.
Επιπλέον, όπως αναφέρθηκε, μεταλλάξεις στο γονίδιο PIK3CA, που συμμετέχει στο μονοπάτι PI3K/AKT/mTOR, παρατηρούνται στο 30% των καρκίνων μαστού και συνδέονται με μειωμένη ενεργοποίηση του μονοπατιού PI3K και ευνοϊκότερη πρόγνωση. Οι μεταλλάξεις αυτές ανευρίσκονται ταυτόχρονα με γενετικές μεταβολές σε άλλα μονοπάτια. Μεταφραστικές έρευνες αρχικώς απέδειξαν το σημαντικό ρόλο αυτού του μονοπατιού. Η μελέτη φάσης ΙΙ TAMRAD (Tamoxifen Plus Everolimus) αξιολόγησε το συνδυασμό του everolimus, ενός αναστολέα mTOR, με ταμοξιφαίνη, σε μετεμμηνοπαυσιακές ασθενείς με HER2(-)/ER(+) μεταστατικούς όγκους μετά από αποτυχία αναστολέα αρωματάσης. Η μελέτη αυτή έδειξε κλινικό όφελος από το συνδυασμό. H αντίστοιχη μελέτη φάσης ΙΙΙ BOLERO-2 (Breast Cancer Trials of Oral Everolimus-2) απέδειξε ότι ο συνδυασμός εξαμεστάνης και everolimus υπερέχει της μονοθεραπείας με εξαμεστάνη σε ασθενείς που υποτροπίασαν μετά από χορήγηση μη στεροειδικού αναστολέα αρωματάσης, και αυτός ο συνδυασμός έχει λάβει την αντίστοιχη έγκριση για χορήγηση από την Ευρωπαϊκή Εταιρεία Έγκρισης Φαρμάκων (Εuropean Medicine Agency-EMA). Τέλος, σε μια μελέτη φάσης ΙΙ, ο συνδυασμός λετροζόλης και everolimus ως προεγχειρητική θεραπεία οδήγησε σε αύξηση της κλινικής ανταπόκρισης, σε συγχορήγηση για 4 μήνες πριν από το χειρουργείο.
Τα δεδομένα αυτά επιδεικνύουν ότι το όφελος του συνδυασμού της ορμονικής θεραπείας με αναστολή των μονοπατιών αυξητικών παραγόντων είναι δυνητικώς μεγαλύτερο σε ασθενείς που αρχικώς απαντούν στην ορμονοθεραπεία και ακολούθως αποκτούν αντίσταση. Αυτό πιθανώς συμβαίνει γιατί εγκαθίσταται η αποκλίνουσα λειτουργία των μονοπατιών και η ικανότητα τους να ανατρέπουν τη σηματοδότηση του ER μπορεί να χρησιμοποιηθεί για θεραπευτική παρέμβαση. Αντιθέτως, ο συνδυασμός δεν αποδεικνύεται ωφέλιμος σε μη επιλεγμένους ασθενείς που δεν έχουν λάβει ορμονοθεραπεία πρώτης γραμμής. Κατά αναλογία με αυτό, ο συνδυασμός λετροζόλης με temsirolimus δεν υπερείχε της λετροζόλης ως πρώτης γραμμής θεραπείας ορμονοευαίσθητων όγκων σε μια φάσης ΙΙ μελέτη.
3. Άλλα μόρια στόχευσης
Άλλωστε, όπως προαναφέρθηκε, οι αναστολείς HDAC έχουν δείξει υποσχόμενα αποτελέσματα σε προκλινικές μελέτες. Σε κλινικό επίπεδο, στη μελέτη ENCORE 301, ο συνδυασμός του αναστολέα HDAC entinostat με εξαμεστάνη είχε ως αποτέλεσμα αύξηση τόσο στην επιβίωση ελεύθερης προόδου νόσου [progression free survival (PFS)-4,3 μήνες έναντι 2,3 μηνών] όσο και στην ολική επιβίωση (26, 9 έναντι 19,9 μηνών).
Η κινάση εξαρτώμενη από την κυκλίνη (cyclin-dependent kinase-CDK) 4/6 αποτελεί σημαντικό παράγοντα ρύθμισης του κυτταρικού κύκλου και η αναστολή του επιφέρει την παύση του κύκλου στη φάση G1. H συγχορήγηση ενός εκλεκτικού αναστολέα CDΚ 4/6 (palbociclib) με τη λετροζόλη έχει βρεθεί ότι οδηγεί σε δραματική αύξηση του PFS (26 έναντι 7,5 μηνών) ως πρώτης γραμμής θεραπεία σε ασθενείς με ΕR θετικό προχωρημένο καρκίνο μαστού. Ως προβλεπτικός βιοδείκτης ελέγχεται η ενίσχυση του γονιδίου της κυκλίνης D1 και η απώλεια p16. Eπιπλέον, τρέχουσες μελέτες αξιολογούν το συνδυασμό αναστολέων αρωματάσης με dasatinib, ενός αναστολέα τυροσινικής κινάσης BCR/Abl. Tέλος η αποτελεσματικότητα pan/PI3K αναστολέων και αναστολέων ΑΚΤ είναι υπό αξιολόγηση σε τρέχουσες κλινικές μελέτες. Είναι φανερό ότι η παρουσία ποικίλλων στοχεύουσων θεραπειών παρέχει τη δυνατότητα προσαρμογής της θεραπείας με φαρμακευτικούς συνδυασμούς και εξατομίκευσης της αντιμετώπισης της αντίστασης στην ορμονική θεραπεία (44-49).
Επίλογος
Συμπερασματικά, η αντίσταση στην ορμονοθεραπεία αποδίδεται σε πολλαπλούς μηχανισμούς. Επειδή ο καρκίνος του μαστού αποτελεί ετερογενή νόσο τόσο σε κλινικό όσο και σε μοριακό επίπεδο, η αντιμετώπιση της αντίστασης πρέπει να εξατομικεύεται. Ο συνδυασμός ορμονοθεραπείας με άλλα φάρμακα που στοχεύουν πρωτεϊνικά μόρια-κλειδιά που εμπλέκονται στην αντίσταση στην ενδοκρινική θεραπεία αποτελεί πολλά υποσχόμενη προσέγγιση για την υπερνίκηση ή την πρόληψη της αντίστασης, με απώτερο στόχο το κλινικό όφελος των ασθενών.
ΒΙΒΛΙΟΓΡΑΦΙΑ
1. Clark GM, Osborne CK, McGuire WL. Correlations between estrogen receptor, progesterone receptor, and patient characteristics in human breast cancer. J Clin Oncol. 1984 Oct;2(10):1102-9.
2. Gradishar WJ. Tamoxifen–what next? Oncologist. 2004;9(4):378-84.
3. Miller WR, Bartlett JM, Canney P, Verrill M. Hormonal therapy for postmenopausal breast cancer: the science of sequencing. Breast Cancer Res Treat. 2007 Jun;103(2):149-60.
4. Osborne CK, Schiff R, Fuqua SA, Shou J. Estrogen receptor: current understanding of its activation and modulation. Clin Cancer Res. 2001 Dec;7(12 Suppl):4338s-42s; discussion 411s-412s.
5. Osborne CK, Schiff R. Estrogen-receptor biology: continuing progress and therapeutic implications. J Clin Oncol. 2005 Mar 10;23(8):1616-22.
6. Mann S, Laucirica R, Carlson N, Younes PS, Ali N, Younes A, et al. Estrogen receptor beta expression in invasive breast cancer. Hum Pathol. 2001 Jan;32(1):113-8.
7. Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998 Nov 26;339(22):1609-18.
8. Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci U S A. 1992 May 1;89(9):4037-41.
9. Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. N Engl J Med. 2003 Jun 12;348(24):2431-42.
10. Parker MG. Action of “pure” antiestrogens in inhibiting estrogen receptor action. Breast Cancer Res Treat. 1993;26(2):131-7.
11. Burstein HJ, Prestrud AA, Seidenfeld J, Anderson H, Buchholz TA, Davidson NE, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol. 2010 Aug 10;28(23):3784-96.
12. Rakha EA, Reis-Filho JS, Ellis IO. Combinatorial biomarker expression in breast cancer. Breast Cancer Res Treat. 2010 Apr;120(2):293-308.
13. Rakha EA, El-Sayed ME, Green AR, Paish EC, Powe DG, Gee J, et al. Biologic and clinical characteristics of breast cancer with single hormone receptor positive phenotype. J Clin Oncol. 2007 Oct 20;25(30):4772-8.
14. Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005 Apr 10;23(11):2469-76.
15. Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994 May 15;54(10):2552-5.
16. Parl FF. Multiple mechanisms of estrogen receptor gene repression contribute to ER-negative breast cancer. Pharmacogenomics J. 2003;3(5):251-3.
17. Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional activation of the estrogen receptor-alpha and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer Res. 2011 Mar 1;71(5):1893-903.
18. Trimarchi MP, Mouangsavanh M, Huang TH. Cancer epigenetics: a perspective on the role of DNA methylation in acquired endocrine resistance. Chin J Cancer. 2011 Nov;30(11):749-56.
19. Lo PK, Sukumar S. Epigenomics and breast cancer. Pharmacogenomics. 2008 Dec;9(12):1879-902.
20. Cui J, Germer K, Wu T, Wang J, Luo J, Wang SC, et al. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer Res. 2012 Nov 1;72(21):5625-34.
21. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003 Mar 5;95(5):353-61.
22. Nicholson RI, Hutcheson IR, Jones HE, Hiscox SE, Giles M, Taylor KM, et al. Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev Endocr Metab Disord. 2007 Sep;8(3):241-53.
23. Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol. 2009 Nov 20;27(33):5529-37.
24. Wang X, Masri S, Phung S, Chen S. The role of amphiregulin in exemestane-resistant breast cancer cells: evidence of an autocrine loop. Cancer Res. 2008 Apr 1;68(7):2259-65.
25. Massarweh S, Osborne CK, Jiang S, Wakeling AE, Rimawi M, Mohsin SK, et al. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006 Aug 15;66(16):8266-73.
26. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997 Mar 28;275(5308):1943-7.
27. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003 Mar;17(3):590-603.
28. Hurvitz SA, Pietras RJ. Rational management of endocrine resistance in breast cancer: a comprehensive review of estrogen receptor biology, treatment options, and future directions. Cancer. 2008 Nov 1;113(9):2385-97.
29. Clarke R, Skaar TC, Bouker KB, Davis N, Lee YR, Welch JN, et al. Molecular and pharmacological aspects of antiestrogen resistance. J Steroid Biochem Mol Biol. 2001 Jan-Mar;76(1-5):71-84.
30. Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst. 2000 Dec 6;92(23):1926-34.
31. Antoon JW, Bratton MR, Guillot LM, Wadsworth S, Salvo VA, Elliott S, et al. Pharmacology and anti-tumor activity of RWJ67657, a novel inhibitor of p38 mitogen activated protein kinase. Am J Cancer Res. 2012;2(4):446-58.
32. Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007;7:59.
33. Chang HL, Sugimoto Y, Liu S, Ye W, Wang LS, Huang YW, et al. Keratinocyte growth factor (KGF) induces tamoxifen (Tam) resistance in human breast cancer MCF-7 cells. Anticancer Res. 2006 May-Jun;26(3A):1773-84.
34. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004 Jan 23;116(2):281-97.
35. Iorio MV, Casalini P, Piovan C, Braccioli L, Tagliabue E. Breast cancer and microRNAs: therapeutic impact. Breast. 2011 Oct;20 Suppl 3:S63-70.
36. Girault I, Lerebours F, Amarir S, Tozlu S, Tubiana-Hulin M, Lidereau R, et al. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin Cancer Res. 2003 Apr;9(4):1259-66.
37. McBryan J, Theissen SM, Byrne C, Hughes E, Cocchiglia S, Sande S, et al. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res. 2012 Jan 15;72(2):548-59.
38. Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006 Mar 13;25(11):1679-91.
39. Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer. 2009 Aug;9(8):576-86.
40. Chia S, Gradishar W, Mauriac L, Bines J, Amant F, Federico M, et al. Double-blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor-positive, advanced breast cancer: results from EFECT. J Clin Oncol. 2008 Apr 1;26(10):1664-70.
41. Dodwell D, Coombes G, Bliss JM, Kilburn LS, Johnston S. Combining fulvestrant (Faslodex) with continued oestrogen suppression in endocrine-sensitive advanced breast cancer: the SoFEA trial. Clin Oncol (R Coll Radiol). 2008 Jun;20(5):321-4.
42. Bergh J, Jonsson PE, Lidbrink EK, Trudeau M, Eiermann W, Brattstrom D, et al. FACT: an open-label randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first-line therapy for patients with receptor-positive postmenopausal breast cancer. J Clin Oncol. 2012 Jun 1;30(16):1919-25.
43. Mehta RS, Barlow WE, Albain KS, Vandenberg TA, Dakhil SR, Tirumali NR, et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med. 2012 Aug 2;367(5):435-44.
44. Smith IE, Walsh G, Skene A, Llombart A, Mayordomo JI, Detre S, et al. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J Clin Oncol. 2007 Sep 1;25(25):3816-22.
45. Johnston S, Pippen J, Jr., Pivot X, Lichinitser M, Sadeghi S, Dieras V, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol. 2009 Nov 20;27(33):5538-46.
46. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224.
47. Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012 Feb 9;366(6):520-9.
48. Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012 Aug 1;30(22):2718-24.
49. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77.






