Από τους
Ευάγγελο Ντουβέλη και Μάριο Μπακογεώργο
Παθολόγους Ογκολόγους
Εισαγωγή
Ο καρκίνος του παχέος εντέρου και του ορθού (ΚΠΕΟ) είναι η τέταρτη πιο συχνή αιτία θανάτου από καρκίνο στις Ηνωμένες Πολιτείες με άνω των 140.000 νέων περιπτώσεων και άνω των 50.000 θανάτων ανά έτος (1). Κατά την αρχική διάγνωση, 40-50% των ασθενών με ΚΠΕΟ έχουν μεταστατική νόσο, γεγονός που υποδηλώνει τη σημασία μίας αποτελεσματικής συστηματικής θεραπείας (2). Κατά τη διάρκεια των τελευταίων 10 ετών, χημειοθεραπευτικοί και βιολογικοί παράγοντες όπως oxaliplatin, irinotecan, cetuximab (CTX), panitumumab (PAM), bevacizumab, aflibercept και regorafenib (3-6), έχουν εγκριθεί ως συμπληρωματικοί στην παραδοσιακή θεραπεία με φθοριοουρακίλη (5 – FU), διπλασιάζοντας την διάμεση συνολική επιβίωση (OS) από τους 12 στους 22 μήνες. Ωστόσο, η εισαγωγή αυτών των παραγόντων έχει αυξήσει σημαντικά το κόστος της θεραπείας, ενώ δυνητικά έχουν και αρκετές σοβαρές παρενέργειες. Oι δύο αυτές παράμετροι έθεσαν το ζήτημα της ανακάλυψης βιοδεικτών με βάση τους μηχανισμούς αντίστασης ως μέθοδο για την επιλογή ασθενών που θα ωφεληθούν από μια συγκεκριμένη θεραπεία. Ο προσδιορισμός μεταλλάξεων στο KRAS ογκογονίδιο ως μηχανισμός αντίστασης στην cetuximab (7-9) και στο panitumumab (10-13), εγκαινίασε την εποχή της εξατομικευμένης θεραπευτικής προσέγγισης στον ορθοκολικό καρκίνο. Περίπου, το 40% των ασθενών με ΚΠΕΟ έχουν KRAS μεταλλάξεις και είναι ανθεκτικοί στη θεραπεία με τους αναστολείς του EGFR. Η απουσία KRAS μεταλλάξεων δεν εγγυάται όφελος από τους αναστολείς του EGFR. Ως εκ τούτου, άλλα μονοπάτια αντίστασης καθώς και άλλοι δυνητικοί προβλεπτικοί παράγοντες απαιτούνται για τον εντοπισμό τόσο των ασθενών που δεν ανταποκρίνονται όσο και αυτών που αναπτύσσουν αντοχή μετά από μία αρχική ανταπόκριση επί απουσίας KRAS μεταλλάξεων στο μεταστατικό ΚΠΕΟ.
Υποδοχέας του επιδερμικού αυξητικού παράγοντα (EGFR)
Ο EGFR έχει ταυτοποιηθεί σε αρκετές μορφές επιθηλιακού καρκίνου όπως στον πλακώδη καρκίνο κεφαλής-τραχήλου, στον ΚΠΕΟ, στον καρκίνο του μαστού, του παγκρέατος, στο μη μικροκυτταρικό καρκίνο του πνεύμονα και στον καρκίνο του εγκεφάλου. Ο EGFR είναι μια γλυκοπρωτεΐνη 170 kDa, που κωδικοποιείται από ένα γονίδιο που βρίσκεται στο χρωμόσωμα 7p12. Ο EGFR είναι μέλος της οικογένειας του ανθρώπινου επιδερμικού υποδοχέα τυροσινικής κινάσης (Her), η οποία αποτελείται από τον EGFR (erbB1/Her1), τον Her2/neu (erbB2), τον Her3 (erbB3) και τον Her4 (erbB4). Ο EGFR έχει μία εξωκυτταρική περιοχή δέσμευσης συνδέτη, μια μεμβρανική περιοχή μονής σύνδεσης, ένα διαμεμβρανικό πυρηνικό σήμα εντοπισμού και μία κυτταροπλασματική περιοχή τυροσινικής κινάσης. Η ενεργοποίηση του EGFR από προσδέτες όπως ο EGF, ο TGFa, η αμφιρεγκουλίνη, ο συνδεόμενος με την ηπαρίνη EGF, η βητασελλουλίνη και η επιρεγκουλίνη οι οποίοι συνδέονται στον αντίστοιχο υποδοχέα, προκαλεί έτερο- ή ομο-διμερισμό και ενεργοποίηση της περιοχής τυροσινικής κινάσης. Τα φωσφορυλιωμένα κυτταροπλασματικά τμήματα χρησιμεύουν ως θέσεις σύνδεσης για πολλές πρωτεΐνες που περιέχουν src ομόλογα τμήματα και περιοχές δέσμευσης φωσφοτυροσίνης. Η ενεργοποίηση του EGFR διεγείρει πολύπλοκες ενδοκυττάριες οδούς μεταγωγής σήματος που ρυθμίζονται στενά από την παρουσία και το είδος του συνδέτη, τη σύνθεση του ετεροδιμερούς και τη διαθεσιμότητα των πρωτεϊνών δέσμευσης φωσφοτυροσίνης. Οι δύο κύριες οδοί μεταγωγής σήματος που ενεργοποιούνται από τον EGFR είναι τα RAS – RAF – ΜΑΡΚ και PI3K-PTEN/PTEN/AKT μονοπάτια (Σχήμα 1) . Η οδός PI3K/AKT, όταν ενεργοποιηθεί, οδηγεί σε αύξηση της πρωτεϊνοσύνθεσης, της κυτταρικής ανάπτυξης, της κινητικότητας και της επιβίωσης. Το μονοπάτι RAS/ RAF/MAPK οδηγεί στην πρόοδο του κυτταρικού κύκλου και στον πολλαπλασιασμό (14,15).
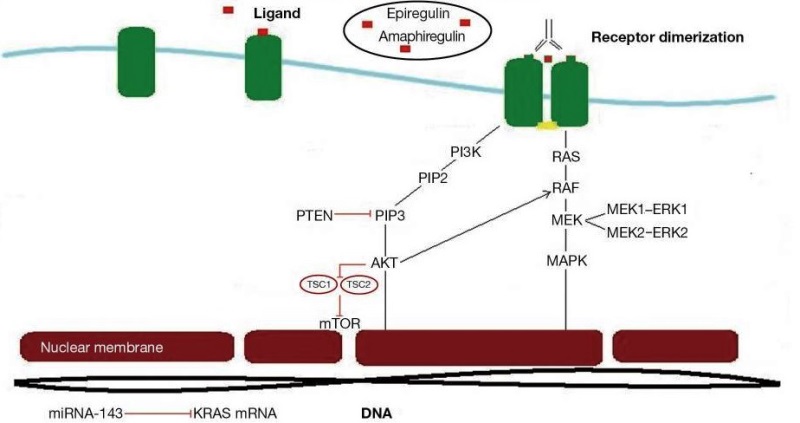
Σχήμα 1: Βιολογική δράση του EGFR. Με κόκκινο χρώμα οι προσδέτες (επιρεγκουλίνη, αμφιρεγκουλίνη) που δεσμεύονται στην εξωκυττάριο περιοχή του EGFR, προκαλούν ομο- ή ετεροδιμερισμό, και οδηγούν σε φωσφορυλίωση των τμημάτων τυροσίνης (κίτρινο) . Η ενεργοποίηση του EGFR οδηγεί στην ενεργοποίηση του RAS, η οποία με τη σειρά της προκαλεί ενεργοποίηση του ογκογονιδίου BRAF, της πρωτεϊνικής κινάσης ΜΕΚ , και της πρωτεΐνης ΜΑΡΚ, οδηγώντας στην έκφραση των γονιδίων που προάγουν την ανάπτυξη. Εκτός από την ενεργοποίηση του RAS, ο EGFR ενεργοποιεί την PIK3CA οποία με τη σειρά , φωσφορυλιώνει την ΡΙΡ2 προς PIP3 και ενεργοποιεί την ΑΚΤ, με αποτέλεσμα τη σύνθεση πρωτεϊνών και την επαγωγή της κυτταρικής ανάπτυξης, της επιβίωσης, του πολλαπλασιασμού, της μετανάστευσης και της αγγειογένεσης. Στους πιθανούς μηχανισμούς αντίστασης περιλαμβάνεται και το miRNA – 143 το οποίο έχει χαμηλή έκφραση στα καρκινικά κύτταρα όγκου και οδηγεί σε μικρότερη αναστολή του KRAS και μεγαλύτερο πολλαπλασιασμό. [Τροποποιημένο από Shaib W και συν (16)].
RAS
KRAS μεταλλάξεις στο εξόνιο 2
Το ανθρώπινο ομόλογο του ογκογονιδίου KRAS, κωδικοποιεί μία μικρή πρωτεΐνη δέσμευσης GTP που δρα ως αυτο-αδρανοποιούμενος μετατροπέας σήματος σε απάντηση στη διέγερση ενός υποδοχέα κυτταρικής επιφάνειας, συμπεριλαμβανομένου του EGFR. Το KRAS μπορεί να φέρει ογκογόνες μεταλλάξεις με αποτέλεσμα την παραγωγή μίας ενεργού πρωτεΐνης. Δεδομένου ότι το KRAS διαδραματίζει βασικό ρόλο στη μεταγωγή του EGFR, έχει αξιολογηθεί η επίδραση των KRAS μεταλλάξεων ως μηχανισμός αντίστασης στην αναστολή του EGFR.
Οι ενεργές KRAS μεταλλάξεις στο κωδικόνιο 12 ανιχνεύονται σε 35%-45% των ασθενών με ΚΠΕΟ (τόσο στην πρωτοπαθή όσο και στις μεταστατικές εστίες αλλά όχι στους λεμφαδένες). Αρκετές αναδρομικές μελέτες έχουν δείξει αντίσταση στους αντι-EGFR στοχευμένους παράγοντες σε ασθενείς των οποίων οι όγκοι φέρουν KRAS μετάλλαξη (6,17). Ο ρόλος των KRAS μεταλλάξεων στην αντοχή στους EGFR αναστολείς καταδεικνύεται καλύτερα σε δύο σημαντικές μελέτες που συγκρίνουν τη μονοθεραπεία με αναστολέα του EGFR σε σχέση με τη βέλτιστη υποστηρικτική φροντίδα. Στην πρώτη μελέτη (NCIC), 572 ασθενείς με χημειοανθεκτική νόσο τυχαιοποιήθηκαν να λάβουν CTX ή την καλύτερη υποστηρικτική φροντίδα (BSC), ως θεραπεία τρίτης γραμμής. Η θεραπεία με cetuximab συσχετίστηκε με μια σημαντική βελτίωση στη συνολική επιβίωση (OS, HR=0,77, P = 0,005) και στην ελεύθερη προόδου νόσου επιβίωση (PFS, HR=0,68, P < 0,001) (5) . Σε μια υποανάλυση, οι ασθενείς χωρίς KRAS μεταλλάξεις (KRAS WT) παρουσίασαν στατιστικά σημαντική βελτίωση στη συνολική επιβίωση (διάμεση τιμή 9,5 μήνες έναντι 4,8 μηνών, HR=0,55, P < 0,001) και στην ελεύθερη προόδου νόσου επιβίωση (διάμεση τιμή 3,7 μήνες έναντι 1,9 μηνών, HR=0,40, Ρ < 0,001). Στους ασθενείς με KRAS μεταλλάξεις δεν αποδείχθηκε όφελος ούτε στη συνολική επιβίωση (HR= 0,98, P=0,89) ούτε στην ελεύθερη προόδου νόσου επιβίωση (HR=0,99, P=0,96) από τη θεραπεία με CTX σε σύγκριση με τη βέλτιστη υποστηρικτική φροντίδα (18). Παρόμοια αποτελέσματα παρατηρήθηκαν και σε μια τυχαιοποιημένη μελέτη στην οποία συγκρίθηκε η θεραπεία με Panitumumab έναντι της βέλτιστης υποστηρικτικής φροντίδας σε ασθενείς με χημειοανθεκτικό μεταστατικό ΚΠΕΟ. Στους ασθενείς χωρίς KRAS μεταλλάξεις καταγράφηκε μια σημαντική βελτίωση στην ελεύθερη προόδου νόσου επιβίωση, με διάμεση τιμή 8 εβδομάδων έναντι 7,3 εβδομάδες στην ομάδα που έλαβε βέλτιστη υποστηρικτική φροντίδα (HR=0,54, P < 0,0001). Τα ποσοστά αντικειμενικής ανταπόκρισης (RR) ήταν υψηλότερα στους ασθενείς που έλαβαν PAM, ωστόσο οι ασθενείς με KRAS μεταλλάξεις δεν είχαν κανένα όφελος ούτε στη συνολική ούτε στην ελεύθερη προόδου νόσου επιβίωση (19).
Στη μελέτη CRYSTAL, 1198 ασθενείς με προχωρημένο ΚΠΕΟ οι οποίοι δεν είχαν λάβει προηγουμένως θεραπεία, τυχαιοποιήθηκαν να λάβουν FOLFIRI ή FOLFIRI + CTX. Διαπιστώθηκε ένα σημαντικό πλεονέκτημα στην ελεύθερη προόδου νόσου επιβίωση στο σκέλος της συνδυασμένης θεραπείας με CTX έναντι του σκέλους που έλαβε μόνο FOLFIRI (HR=0,85, P=0,048), ενώ δεν υπήρχε σημαντική διαφορά στην συνολική επιβίωση (HR=0,93, Ρ=0,31). Σε μια ανάλυση υποομάδας, οι ασθενείς με KRAS μετάλλαξη (37%), δεν είχαν καμία βελτίωση στο διάστημα ελεύθερο προόδου νόσου (HR= 1,07, Ρ=0,75) ή στη συνολική επιβίωση (HR=1,03), με την προσθήκη CTX στη χημειοθεραπεία. Ωστόσο, οι KRAS WT ασθενείς είχαν μία στατιστικά σημαντική βελτίωση τόσο στο διάστημα ελεύθερο προόδου νόσου με ένα μέσο όρο 9,9 μηνών έναντι 8,4 μήνες στην ομάδα της θεραπείας με FOLFIRI (HR=0,68, Ρ=0,02), όσο και στη συνολική επιβίωση (διάμεση διάρκεια 23,5 μήνες έναντι 20 μηνών, HR=0,84, P=0,0093) υπέρ της προσθήκης CTX. Οι αντικειμενικές ανταποκρίσεις ήταν 57,3 % έναντι 39,7 % (P = 0,001) υπέρ του σκέλους που έλαβε και CTX. Τα αποτελέσματα αυτής της μελέτης οδήγησαν στην έγκριση του CTX στη θεραπεία πρώτης γραμμής σε συνδυασμό με FOLFIRI σε KRAS WT ασθενείς με μεταστατικό ΚΠΕΟ ( 6,20 ).
Ενεργές μεταλλάξεις στο κωδικόνιο 13 του γονιδίου KRAS υπάρχουν στο 6 % περίπου των ασθενών με ΚΠΕΟ. Ο ρόλος των μεταλλάξεων του κωδικονίου 13 στην ανάπτυξη αντίστασης στην θεραπεία με EGFR αναστολείς είναι ακόμα αμφιλεγόμενη. Μια in vitro μελέτη έδειξε ότι οι KRAS μεταλλάξεις στο κωδικόνιο 13 εμφανίζουν ασθενέστερη δραστηριότητα μετασχηματισμού από ό, τι οι μεταλλάξεις στο κωδικόνιο 12, με χαμηλή αντίσταση στην απόπτωση και την ικανότητα ανάπτυξης (21) . Οι DeRoock και συν. μελέτησαν τη σχέση μεταξύ της p.G13D μετάλλαξης στην ανταπόκριση και την επιβίωση σε ασθενείς με χημειοανθεκτικό ΚΠΕΟ που λαμβάνουν θεραπεία με CTX. Οι ασθενείς με G13D μεταλλαγμένους όγκους είχαν μεγαλύτερη συνολική επιβίωση (7,6 μήνες έναντι 5,7 μηνών, P=0,005) και διάστημα ελεύθερο προόδου νόσου (4 έναντι 1,9 μήνες, P=0,004). Παρόλο που αυτά τα αποτελέσματα υποδεικνύουν ότι οι ασθενείς με p.G13D μεταλλάξεις ανταποκρίνονται στη θεραπεία με CTX, τα αποτελέσματα έδειξαν χαμηλότερα ποσοστά αντικειμενικών ανταποκρίσεων σε σχέση με τους ασθενείς με KRAS WT όγκους. Από την ίδια μελέτη, ανάλυση in vitro και σε μοντέλα ποντικών έδειξε ότι τα p.G12V μεταλλαγμένα κύτταρα ΚΠΕΟ ήταν αδρανή ενώ τα p.G13D μεταλλαγμένα κύτταρα ήταν ευαίσθητα (όπως τα KRAS WT κύτταρα), στο CTX (22). Οι Peeters και συν. αξιολόγησαν την επίδραση της KRAS μετάλλαξης στο κωδικόνιο 13 σε τρεις μελέτες που διερεύνησαν την αποτελεσματικότητα της χορήγησης PAM σε ασθενείς με προχωρημένο στάδιο ΚΠΕΟ. Τα αποτελέσματα έδειξαν ότι οι ασθενείς με KRAS μετάλλαξη στο κωδικόνιο 13 δεν ωφελούνται από το PAM. Πιθανές ερμηνείες για τη διαφορά στην επίδραση της KRAS μετάλλαξης στο κωδικόνιο 13 αναφορικά με την ευαισθησία στους αναστολείς του EGFR μπορούν να περιλαμβάνουν είτε διαφορά μεταξύ των ΡΑΜ και CTX ως φάρμακα ή πιο πιθανό μια διαφορά στην αλληλεπίδραση του μεταλλαγμένου κωδικονίου 13 με τη χημειοθεραπεία. Πάντως, η απουσία μεγάλων προοπτικών μελετών και τα αντιφατικά αποτελέσματα των δύο αναδρομικών ερευνών , καταδεικνύουν ότι ο ρόλος της μετάλλαξης του κωδικονίου 13 του KRAS στην αντίσταση στoυς αναστολείς του EGFR είναι ακόμα αμφιλεγόμενη (23).
NRAS και λοιπές KRAS μεταλλάξεις
Στη μεγάλη τυχαιοποιημένη μελέτη PRIME που συνέκρινε ασθενείς με προχωρημένο ΚΠΕΟ που έλαβαν FOLFOX με ή χωρίς PAM, 17% των ασθενών χωρίς μεταλλάξεις στο εξόνιο 2 του KRAS βρέθηκαν να έχουν μεταλλάξεις στα εξόνια 3 και 4 του KRAS ή στα εξόνια 2, 3 και 4 του NRAS (24). Σε μία αναδρομική υποανάλυση, τόσο η συνολική επιβίωση όσο και το διάστημα ελεύθερο προόδου νόσου ήταν ελάττωμένα στους ασθενείς με οποιαδήποτε KRAS ή NRAS μετάλλαξη που έλαβαν και PAM σε σχέση με αυτούς που έλαβαν μόνο FOLFOX.
Στην επικαιροποιημένη ανάλυση της μελέτης FIRE-3, οι ασθενείς με οποιαδήποτε RAS (KRAS/NRAS) μετάλλαξη που έλαβαν FOLFIRI/CTX είχαν χειρότερο PFS σε σχέση με όσους είχαν λάβει FOLFIRI/Bevacizumab (25). Επιπλέον, οι KRAS/NRAS WT ασθενείς δεν παρουσίασαν διαφορά στο PFS. Φαίνεται επομένως ότι το CTX έχει αρνητική επίδραση στους ασθενείς με οποιαδήποτε RAS (KRAS/NRAS) μετάλλαξη.
Έτσι ο FDA πρόσφατα επικαιροποίησε τις ενδείξεις για το PAM, προβλέποντας ότι αντενδείκνυται η χορήγησή του με σχήματα που περιέχουν Oxaliplatin σε ασθενείς με (KRAS/NRAS) μετάλλαξη, ενώ πλέον και οι συστάσεις του NCCN είναι να προσδιορίζονται όλες οι RAS μεταλλάξεις και στους ασθενείς με οποιαδήποτεανευρεθείσα KRAS ή NRAS μετάλλαξη να μη χορηγείτα θεραπεία με CTX ή PAM.
Μηχανισμοί αντίστασης πέρα από το RAS
Περίπου οι μισοί από τους ασθενείς χωρίς KRAS/NRAS μεταλλάξεις δεν ανταποκρίνονται στην αντι – EGFR θεραπεία, θέτοντας το ερώτημα για το ποιοί παράγοντες πέραν της κατάστασης των KRAS/NRAS επηρεάζουν την αντίσταση. Οι πιθανοί παράγοντες περιλαμβάνουν την αυξημένη έκφραση του συνδέτη του EGFR, τη μειωμένη έκφραση EGFR, ή την ενεργοποίηση εναλλακτικών οδών μεταγωγής σήματος.
Επίπεδο έκφρασης του EGFR, επιρεγκουλίνη και αμφιρεγκουλίνη
Οι Baker και συν. μελέτησαν βιοπτικό υλικό από πρωτογενείς εστίες αναφορικά με τα επίπεδα γονιδιακής έκφρασης των συνδετών του KRAS και του EGFR. Μεταλλάξεις KRAS βρέθηκαν στο 43 % των ασθενών. Στην ομάδα KRAS WT, η ευαισθησία στην αναστολή του EGFR ήταν ανάλογη με την έκφραση των συνδετών του EGFR, επιρεγκουλίνη και αμφιρεγκουλίνη. Η υψηλή έκφραση συνδέτη εντόπισε μια υποομάδα ασθενών KRAS WT που είχαν μεγάλη πιθανότητα να ανταποκριθούν σε αντι-EGFR θεραπεία σε σύγκριση με τους ασθενείς KRAS WT με χαμηλή έκφραση συνδέτη που συμπεριφέρονταν σαν ασθενείς με μεταλλαγμένο KRAS. Επιπλέον, οι ασθενείς με υψηλά επίπεδα των συνδετών του EGFR ήταν πιο πιθανό να επιτύχουν έλεγχο της νόσου με CTX και σημαντικά μεγαλύτερο διάστημα ελεύθερο προόδου νόσου σε σχέση με τους ασθενείς με χαμηλή έκφραση τόσο επιρεγκουλίνης (Ρ = 0,0002) όσο και αμφιρεγκουλίνης (Ρ = 0,0001) (26). Δεν υπήρξε καμία ένδειξη συσχέτισης μεταξύ της έκφρασης των γονιδίων επιρεγκουλίνης και αμφιρεγκουλίνης και του διαστήματος ελεύθερου προόδου νόσου ή της συνολικής επιβίωσης σε ασθενείς με KRAS μεταλλάξεις (27). Σε ασθενείς με υψηλά επίπεδα mRNA για επιρεγκουλίνη και αμφιρεγκουλίνη, η θεραπεία CTX τείνει να έχει μια πιο ισχυρή αντικαρκινική δραστικότητα. Ως εκ τούτου, η χαμηλή έκφραση του συνδέτη μπορεί να είναι ένας μηχανισμός αντίστασης στους αναστολείς EGFR, καθώς δείχνει ότι το σύστημα του EGFR μπορεί να μην είναι ο κύριος συντελεστής της ανάπτυξης ή της εξέλιξης του όγκου.
Η έκφραση του EGFR
Στην αρχική ανάπτυξη των αναστολέων EGFR, οι ασθενείς επελέγησαν να περιληφθούν σε κλινικές μελέτες μόνο αν οι όγκοι ήταν θετικοί για έκφραση του EGFR με τη χρήση ανοσοϊστοχημείας, λόγω της ιδέας ότι η έλλειψη της έκφρασης EGFR, οδηγεί σε αντοχή σε αναστολείς EGFR. Στην μελέτη των Chung και συν, 4 από τους 16 ασθενείς χωρίς έκφραση EGFR, παρουσίασαν σημαντικές ανταποκρίσεις στη θεραπεία με βάση το CTX (28). Ως εκ τούτου, ο προσδιορισμός της έκφρασης του EGFR με τη χρήση ανοσοϊστοχημείας δεν φαίνεται να επηρεάζει την αντίσταση στους αναστολείς του EGFR.
Ο ρόλος της έκφρασης EGFR στην αντίσταση στους αναστολείς του EGFR αξιολογήθηκε επίσης και σε μοριακό επίπεδο. Σε μία μελέτη των Moroni και συν, 31 ασθενείς με μεταστατικό ΚΠΕΟ που είτε είχαν ανταπόκριση ή σταθεροποίηση νόσου (30%) ή πρόοδο νόσου (70%) μετά από θεραπεία με CTX ή ΡΑΜ, μελετήθηκαν αναφορικά με των αριθμό αντιγράφων του γονιδίου του EGFR. Οκτώ από τους εννέα ασθενείς με αντικειμενικές ανταποκρίσεις είχαν αυξημένο αριθμό αντιγράφων του EGFR. Από την άλλη πλευρά, ένας από τους 21 μη ανταποκρινόμενους ασθενείς είχαν αυξημένο αριθμό αντιγράφων του EGFR (Ρ < 0,0001 ) (29). Η ίδια ομάδα αξιολόγησε τον ρόλο του αριθμού αντιγράφων του EGFR ως προγνωστικό δείκτη της κλινικής έκβασης σε ασθενείς που έλαβαν θεραπεία με το PAM . Ένας μέσος αριθμός αντιγράφων του γονιδίου EGFR μικρότερος από 2,5/πυρήνα ή μικρότερος από το 40% των κυττάρων του όγκου που εμφανίζουν πολυσωμία του χρωμοσώματος 7 εντός του όγκου, κατεδείχθη προβλεπτικός για μικρότερο διάστημα ελεύθερο προόδου νόσου ( Ρ = 0,039) και χειρότερη συνολική επιβίωση (Ρ=0,015) (30). Οι Lenz και συν. αξιολόγησαν επίσης την επίδραση του αριθμού αντιγράφων του γονιδίου EGFR στην ανταπόκριση στο CTX χρησιμοποιώντας αλυσιδωτή αντίδραση πολυμεράσης (PCR), αντί του φθορίζοντος in situ υβριδισμού (FISH) που είχε χρησιμοποιηθεί στις προηγούμενες μελέτες. Παρατηρήθηκε έλλειψη συσχέτισης του αυξημένου αριθμού αντιγράφων του γονιδίου με τις αντικειμενικές ανταποκρίσεις και την ελεύθερη προόδου νόσου επιβίωση, ωστόσο καταδείχθηκε θετική συσχέτιση με τη συνολική επιβίωση (31). Η αναδρομική ανάλυση αριθμού αντιγράφων EGFR με FISH από 85 δείγματα χημειοανθεκτικών ασθενών με μεταστατικό ΚΠΕΟ που έλαβαν θεραπεία με CTX, ανίχνευσε το θετικό EGFR FISH score που συσχετίζεται καλύτερα με την αντικειμενική ανταπόκριση και την επιμήκυνση του διαστήματος ως την πρόοδο της νόσου σε σύγκριση με τo αρνητικό EGFR FISH score σε μια μέση τιμή 2,92 αριθμού αντιγράφων γονιδίου EGFR (32, 33). Στη μελέτη των Lievre και συν, ο αυξημένος αριθμός αντιγράφων του γονιδίου EGFR που μετρήθηκε με χρωμογόνο in situ υβριδισμό (CISH) συσχετίστηκε σημαντικά με την αντικειμενική ανταπόκριση του όγκου στη θεραπεία με CTX, όμως ο μικρός αριθμός των EGFR-θετικών ασθενών δεν επιτρέπει οριστικά συμπεράσματα (34). Η μεγαλύτερη έρευνα προς την κατεύθυνση αυτή, ανίχνευσε αυξημένο αριθμό αντιγράφων του γονιδίου EGFR μόνο στο 6% και χωρίς συσχέτιση με το ποσοστό ελέγχου της νόσου (29). Μια πρόσφατη μετα-ανάλυση δείχνει ότι ο αυξημένος αριθμός των αντιγράφων του γονιδίου του EFGR συσχετίζεται με αύξηση της επιβίωσης μετά από αντι-EGFR θεραπεία σε τους ασθενείς με μεταστατικό ΚΠΕΟ (35). Συνολικά, τα τρέχοντα δεδομένα σχετικά με το ρόλο των αριθμών αντιγράφων του γονιδίου EGFR ως μηχανισμός αντίστασης στην αναστολή του EGFR είναι ελλειπή, λόγω τεχνικών σφαλμάτων, μη καθορισμένου ορίου και έλλειψης τυποποίησης. Με τις διαφορετικές μεθόδους που χρησιμοποιούνται ( FISH, qPCR, ή CISH ), είναι δύσκολο να υπάρξει σύγκριση μεταξύ των μελετών.
BRAF
Η κινάση σερίνης – θρεονίνης BRAF είναι ο κύριος παράγοντας που επιδρά στο KRAS. Η μετάλλαξη BRAF συμβαίνει σε κατωφερέστερο του KRAS μονοπάτι και βρίσκεται σε λιγότερο από το 10 % των ασθενών με ΚΠΕΟ. Η συνολική επιβίωση διαφέρει ανάλογα με τις σωματικές μεταλλάξεις, ανεξαρτήτως του είδους της λαμβανόμενης θεραπείας: Επί υπάρξεως BRAF μεταλλάξεων 8,8 μήνες, σε KRAS μεταλλάξεις 14,4 μήνες και σε KRAS WT 20,1 μήνες (36). Η μετάλλαξη V600E στο BRAF συσχετίστηκε με κακή πρόγνωση σε ασθενείς με KRAS WT που έλαβαν είτε FOLFIRI μόνο είτε συνδυασμό FOLFIRI/CTX. Τα άτομα με μεταλλάξεις στο BRAF είχαν χειρότερα αποτελέσματα. Μεταλλάξεις V600E στο BRAF ανιχνεύθηκαν σε 6 % των δειγμάτων. Σε όλες σχεδόν τις περιπτώσεις, αυτές οι μεταλλάξεις εντοπίστηκαν στους KRAS WT όγκους και η επίδραση της BRAF μετάλλαξης σε σχέση με την αποτελεσματικότητα των αντι – EGFR παραγόντων εξετάστηκε στον πληθυσμό της μελέτης CRYSTAL. Η παρουσία της μετάλλαξης BRAF ήταν πτωχός προβλεπτικός δείκτης ανταπόκρισης στη θεραπεία και επιβίωσης. Το αν ο βιοδείκτης αυτός έχει αρνητική προβλεπτική αξία αναφορικά με τη θεραπεία με CTX είναι δύσκολο να προσδιοριστεί αφού αυτή η μελέτη είχε ένα σχετικά μικρό αριθμό ασθενών με μεταλλάξεις BRAF (6). Σε άλλες μελέτες , η BRAF μετάλλαξη ήταν αρνητικός προγνωστικός δείκτης για τη συνολική επιβίωση ασθενών με μεταστατικό ΚΠΕΟ (37, 38) . Στη μελέτη NORDIC VII, οι ασθενείς με μεταλλαγμένο BRAF είχαν χαμηλό ποσοστό αντικειμενικών ανταποκρίσεων και σημαντικά μικρότερη επιβίωση ελεύθερης προόδου νόσου και συνολική επιβίωση σε σύγκριση με εκείνους χωρίς μεταλλάξεις (39). Σε μια αναδρομική μελέτη 113 ασθενών με μεταστατικό ΚΠΕΟ που έλαβαν θεραπεία με CTX ή PAM, καταληκτικά σημεία ήταν το ποσοστό ανταπόκρισης, ο χρόνος έως την εξέλιξη της νόσου, η συνολική επιβίωση και το status των KRAS και BRAF μεταλλάξεων. Η μετάλλαξη V600E του BRAF ανιχνεύθηκε στο 14 % των ασθενών που είχαν νόσο KRAS WT. Κανένας από τους ασθενείς με BRAF μετάλλαξη δεν ανταποκρίθηκε στην αντι – EGFR θεραπεία και είχαν σημαντικά μικρότερη επιβίωση ελεύθερης προόδου νόσου και συνολική επιβίωση σε σύγκριση με τους BRAF WT. Ο ρόλος των BRAF μεταλλάξεων σε ασθενείς που έλαβαν θεραπεία με φάρμακα που στοχεύουν τον EGFR είναι παρόμοιος με εκείνο των KRAS μεταλλάξεων (40). Επιπλέον, το 50 % των μεταλλάξεων BRAF ανιχνεύονται πιο συχνά σε ορθοκολικούς όγκους με υψηλή μικροδορυφορική αστάθεια (41-43). Ακόμη και με την αναστολή του BRAF από το vemurafenib , έχει φανεί περιορισμένη ανταπόκριση. Έχει διατυπωθεί η θεωρία ότι με αυτήν την αναστολή, θα συμβεί η μεγαλύτερη ενεργοποίηση του EGFR, σε αντίθεση με τα κύτταρα του μελανώματος που εκφράζουν χαμηλά επίπεδα του EGFR στην επιφάνειά τους (44-48). Η ανάλυση μίας μελέτης κατά την οποία προστέθηκε sorafenib σε ένα αντι – EGFR παράγοντα έδειξε ότι ακόμη και BRAF μεταλλαγμένα κύτταρα ΚΠΕΟ μπορεί δυνητικά να ανταποκριθούν σε θεραπείες που στοχεύουν τον EGFR, εάν ο BRAF αναστολέας sorafenib χορηγείται ταυτόχρονα με CTX ή PAM , ακόμα και όταν τα δύο αυτά φάρμακα ξεχωριστά έχουν περιορισμένη δραστικότητα. Αυτά τα δεδομένα υποδεικνύουν ότι σε όγκους με BRAF μεταλλάξεις, η θεραπευτική επίδραση του CTX ή ΡΑΜ θα μπορούσε να αποκατασταθεί στοχεύοντας την παρεμπόδιση της οδού του EGFR σε πολλαπλές θέσεις. Εκτός από το sorafenib , άλλες ουσίες που στοχεύουν είτε το BRAF (PLX4032) ή κατωφερέστερα μόρια (ARRY-162, AZD6244 και PD0325901) θα μπορούσαν να αξιοποιηθούν σε συνδυασμό με μία EGFR – στοχευμένη θεραπεία με μονοκλωνικά αντισώματα (49, 50). Παρά την KRAS και BRAF WT κατάσταση, υπάρχει ακόμη ένα σημαντικό ποσοστό μη ανταποκρινόμενων ασθενών (41%) σε αντι – EGFR θεραπείες, θέτοντας το ερώτημα για την ύπαρξη περαιτέρω μονοπατιών σημαντικών για τον καθορισμό της αντίστασης σε αυτές τις θεραπείες (40) .
Γονίδιο PIK3K
Σε μια μελέτη που εξετάζει χημειοανθεκτικούς ασθενείς με ορθοκολικό καρκίνο που αντιμετωπίστηκαν με CTX και χημειοθεραπεία, τα άτομα με PIK3CA μεταλλάξεις στο εξόνιο 20 του KRAS είχαν χειρότερη έκβαση σε σύγκριση με τους KRAS WT, παρουσιάζοντας χαμηλότερο ποσοστό ανταπόκρισης και μειωμένη διάμεση επιβίωση. Οι μεταλλάξεις στο εξώνιο 9 του PIK3CA δεν είχαν καμία επίδραση στην επιβίωση και την πρόγνωση (36) . Παρόμοια ευρήματα παρατηρήθηκαν σε μία ανασκόπηση της συσχέτισης μεταξύ των μεταλλάξεων PIK3CA και της κλινικής έκβασης των ασθενών με μεταστατικό ΚΠΕΟ που υποβλήθηκαν σε θεραπεία με αντι-EGFR μονοκλωνικά αντισώματα. Τα αποτελέσματα αυτά δείχνουν επίσης ότι το εξόνιο 20 του PIK3CA μπορεί να αποτελεί έναν πιθανό βιοδείκτη αντίστασης σε αντι – EGFR μονοκλωνικά αντισώματα σε ασθενείς με KRAS WT (51). Οι μεταλλάξεις στο PIK3CA έχουν συσχετιστεί με αντοχή στην αντι – EGFR θεραπεία , δεδομένου ότι μπορούν να συνυπάρχουν με μεταλλάξεις του γονιδίου KRAS. Ωστόσο, ήταν δύσκολο να επιβεβαιωθεί μία οριστική συσχέτιση. Hot-spot μεταλλάξεις σε στο PIK3CA, μπορεί να λειτουργούν με διαφορετικούς αλλά συνεργιστικούς μηχανισμούς, ανεξάρτητους από το KRAS (52). Ωστόσο, ο ρόλος της μετάλλαξης PIK3CA στην αντίσταση EGFR σε ασθενείς με μεταστατικό ΚΠΕΟ παραμένει αμφιλεγόμενη .
Σε μια μελέτη του PIK3CA σε μια ομάδα 200 χημειοανθεκτικών ασθενών με μεταστατικό ΚΠΕΟ που έλαβαν αγωγή με CTX, οι ασθενείς KRAS WT δεν είχαν καμία διαφορά στην ανταπόκριση στο CTX σε σχέση με την κατάσταση του PIK3CA (μεταλλαγμένο ή μη) (53) . Μεταλλάξεις στο PIK3CA ανιχνεύθηκαν σε 16,4%. Σε μονοπαραγοντική ανάλυση, προγνωστική σημασία για την επιβίωση παρατηρήθηκε για BRAF μεταλλάξεις μόνο επί μεταλλαξεων στο κωδικόνιο 12 του KRAS, για υψηλή έκφραση του mRNA της αμφιρεγκουλίνης μόνο σε KRAS WT ασθενείς και για υψηλή έκφραση του mRNA της επιρεγκουλίνης ανεξάρτητα από την κατάσταση μετάλλαξης του KRAS. Ευνοϊκοί προγνωστικοί παράγοντες ήταν η υψηλή έκφραση mRNA της αμφιρεγκουλίνης σε KRAS WT όγκους , η υψηλή έκφραση mRNA της επιρεγκουλίνης και η χαμηλή έκφραση mRNA του υποδοχέα Ephrin Α2. Οι ασθενείς που έλαβαν θεραπεία με CTX και είχαν χαμηλή αμφιρεγκουλίνη και ήταν KRAS WT είχαν πτωχή ανταπόκριση, με επιβίωση παρόμοια με τους έχοντες KRAS μετάλλαξη . Ασθενείς με KRAS μετάλλαξη στο κωδικόνιο 13 ή άλλες μεταλλάξεις πλην του κωδικονίου 12, είχαν διάμεση επιβίωση παρόμοια με εκείνη των ασθενών με KRAS WT, γεγονός που έρχεται σε αντίθεση με τους ασθενείς με KRAS μεταλλάξεις στο κωδικόνιο 12 που είχαν χειρότερη έκβαση από όλους τους άλλους (54).
Από την άποψη των στοχευμένων θεραπευτικών προσεγγίσεων, οι KRAS μεταλλάξεις παρουσιάζουν ενδείξεις αντοχής στους αναστολείς του μονοπατιού Ρ13Κ (55) . Συγκεκριμένα, η παρουσία των μεταλλαγμένου KRAS ήταν προβλεπτικός δείκτης αντίστασης στον αναστολέα Ρ13Κ , PX- 866 (56). Αυτό μπορεί να περιορίσει τη χρησιμότητα της μονοθεραπείας με αναστολείς της οδού Ρ13Κ που έχουν KRAS και PIK3CA μεταλλάξεις που εμφανίζονται σε καρκίνους του παχέος εντέρου (57).
PTEN
Η ενίσχυση της μεταγωγής σήματος του Ρ13Κ συχνά οφείλεται στην ενεργοποίηση των γονιδίων που εμπλέκονται στο μονοπάτι Ρ13Κ όπως τα PIK3CA και AKT1, ή στην απώλεια του ΡΤΕΝ (58-60). Μεταλλάξεις του PTEN παρατηρήθηκαν σε περίπου 18 % των ασθενών με ορθοκολικούς όγκους που είχαν μικροδορυφορική αστάθεια, γεγονός που υποδηλώνει ότι η ελαττωματική επιδιορθωτική ικανότητα του PTEN μπορεί να είναι ένας πιθανός στόχος για τις μελλοντικές θεραπείες (61, 62) . Πρόσθετα στοιχεία δείχνουν ότι η υπερμεθυλίωση του υποκινητή του PTEN παρουσιάζεται συχνά στους όγκους με υψηλή έναντι χαμηλής μικροδορυφορικής αστάθειας (19,1% έναντι 2,2 %, Ρ = 0,002) (63). Μία συνδυασμένη ανάλυση των KRAS, BRAF και PTEN έδειξε αυξημένη συχνότητα ανταποκρίσεων σε ποσοστό έως 45% για χημειοανθεκτικούς ασθενείς που λαμβάνουν CTX σε αντίθεση με 39% σε ασθενείς με KRAS WT, BRAF WT και PTEN WT όγκους, ενώ οι ασθενείς με PTEN μεταλλάξεις ήταν όλοι ανθεκτικοί στο CTX , σε αντίθεση με τους ασθενείς με KRAS μεταλλάξεις, από τους οποίους το 12,5 % σε αυτή τη μελέτη ανταποκρίθηκαν στη θεραπεία με CTX (64).
ΜΑΡΚ
Η διασταύρωση των KRAS – ΜΑΡΚ – PI3KCA μονοπατιών έχει άμεσες επιπτώσεις για την ογκογένεση. Το ποσοστό των KRAS μεταλλάξεων προσδιορίστηκε με ανάλυση αλληλουχίας του εξωνίου 2 , το οποίο έχει τα πιο συχνά μεταλλαγμένα κωδικόνια (κωδικόνιο 12 και 13) (65). Μία γενετική παραλλαγή στο μονοπάτι σηματοδότησης ΜΑΡΚ επιδρά στον καρκίνο του παχέος εντέρου και μπορεί να επηρεάζεται από περιβαλλοντικούς παράγοντες και τον τρόπο ζωής, συμπεριλαμβανομένης της χρήσης της ασπιρίνης ή άλλων μη στερινοειδών αντιφλεγμονωδών, του καπνίσματος, της έκθεσης σε οιστρογόνα και του δείκτη μάζας σώματος (66). Ο συνδυασμός της αναστολής των Ρ13Κ και ΜΑΡΚ μονοπατιών με τη χορήγηση ενός διπλού αναστολέα PI3K/mTOR ( NVP – BEZ235 ) και ενός αναστολέα ΜΕΚ ( ARRY – 142886 ) οδήγησε σε σημαντική υποχώρηση του όγκου σε ένα KRAS μοντέλο καρκίνου του πνεύμονα (55) .
ΜΕΚ
Ένα άλλο μόριο-στόχος κατωφερέστερα του KRAS είναι ο MEK, που ενεργοποιεί τις εξωκυττάριες σηματοεπαγώμενες κινάσες (ERK-1 και ERK-2) οι οποίες είναι υπεύθυνες για τη φωσφορυλίωση των παραγόντων που ελέγχουν την ενεργοποίηση του κυτταρικού κύκλου, κυρίως στη μετάβαση από τη φάση G στη φάση S. Αντίσταση στην EGFR στοχευμένη θεραπεία θα μπορούσε επίσης να προκαλείται μέσω εναλλακτικής ενεργοποίησης των ERK-1 και ERK-2 που παρακάμπτει τον EGFR είτε μέσω εναλλακτικών υποδοχέων στην πλασματική μεμβράνη είτε μέσω ενεργών κατωφερέστερων συστατικών. Δημιουργώντας CTX-ανθεκτικές κυτταρικές σειρές, οι Yonesaka και συν. πρώτοι εντόπισαν πολλαπλούς κλώνους που εμφάνισαν λιγότερο αποτελεσματική καταστολή της φωσφορυλίωσης των ERK1 / 2 παρουσία CTX. Περαιτέρω ανάλυση αυτών των κλώνων απεκάλυψε ενίσχυση του ErbB2 με αντίστοιχες αυξήσεις των συνολικών και των φωσφο-ΕrbΒ2 επιπέδων. Επακόλουθη ελάττωση του ErbB2 στους ανθεκτικούς κλώνους αποκαθιστά την ευαισθησία στο CTX, επιβεβαιώνοντας την σημασία του ErbB2 στον ανθεκτικό φαινότυπο. Η ενίσχυση του ErbB2 είναι ο προτεινόμενος μηχανισμός των ανθεκτικών στο CTX κλώνων όπου η αποκτηθείσα αντίσταση προκαλείται από τα αυξημένα επίπεδα της ερεγκουλίνης που συνδέει ErbB3 και ErbB4. Αυτό οδηγεί στην ενεργοποίηση μορίων σε κατωφερέστερα μονοπάτια και ο ρόλος αυτού του προσδέτη δεν έχει ακόμη προσδιοριστεί (67). Σε μια πρόσφατη μοριακή ανάλυση, μοριακές αλλαγές στο γονίδιο KRAS οδήγησαν σε επίκτητη αντίσταση στην αντι – EFGR θεραπεία. Μεταλλαγμένα αλληλόμορφα KRAS σε ασθενείς που έλαβαν θεραπεία με CTX ήταν ανιχνεύσιμα δέκα μήνες πριν από την ακτινολογική ένδειξη εξέλιξης της νόσου . Όταν συνδυάζεται νωρίς ένας αναστολέας EGFR με έναν αναστολέα ΜΕΚ, τα στοιχεία δείχνουν καθυστέρηση ή αναστροφή της φαρμακευτικής αντίστασης (68) .
IGF1
Ο υποδοχέας του IGF ( IGF-1R ) είναι ένα μέλος μιας οικογένειας διαμεμβρανικών τυροσινικών κινασών που περιλαμβάνει τον υποδοχέα της ινσουλίνης και του υποδοχέα που σχετίζεται με τον υποδοχέα ινσουλίνης . Το σηματοδοτικό μονοπάτι του IGF -1R είναι σημαντικό σε διαφορετικούς τύπους καρκίνων και περιλαμβάνει μεταγωγή του IGF σήματος από τον ΜΑΡΚ και PI3K/AKT. Προκλινικά δεδομένα δείχνουν ότι η θεραπεία συνδυασμού IGF-1R και EGFR αναστολέων κινάσης έχουν συνεργική αποτελεσματικότητα στην αναστολή της ανάπτυξης κυτταρικών σειρών ορθοκολικού καρκίνου (69). Ειδικότερα, ο κατωφερέστερος καταρράκτης μεταγωγής σήματος του IGF – 1 πιστεύεται ότι επάγει την ανεξάρτητη από τον EGFR δραστικότητα των PIK3CA και ΑΚΤ, θεωρία που μπορεί να αποτελεί μια άλλη εξήγηση για την έλλειψη αποτελεσματικότητας των αντι- EGFR μονοκλωνικών αντισωμάτων σε KRAS WT ασθενείς με ΚΠΕΟ (70). Το ίδιο υποστηρίζουν οι Bohula και συν. που έδειξαν με τα πειράματά τους ότι ο IGF – 1 και ο IGF – 2 αλληλεπιδρούν με το IGF- 1 υποδοχέα ( IGF1R), ρυθμίζοντας την ανάπτυξη, τη διαφοροποίηση και την επιβίωση των κυττάρων. Ο IGF1R ενεργοποιεί τα RAS/ERK- και PI3K/AKT σχετιζόμενα μονοπάτια μεταγωγής σήματος, που δρουν για να προάγουν τον πολλαπλασιασμό και την πρόληψη της απόπτωσης (71). Μια μελέτη φάσης II με το αντι- IGF-1R μονοκλωνικό αντίσωμα IMC-Α12, είτε μόνο είτε σε συνδυασμό με CTX, διεξήχθη σε ασθενείς με CTX ή PAM- ανθεκτική νόσο. Καμία δραστικότητα κατά του όγκου δεν παρατηρήθηκε στους 23 ασθενείς που έλαβαν μονοθεραπεία με IMC-A12 και τους 21 ασθενείς που έλαβαν θεραπεία με τον συνδυασμό IMC-Α12 και CTX, ενώ ένας ασθενής με KRAS WT είχε μερική ανταπόκριση, με τον έλεγχο της νόσου να διαρκεί 6,5 μήνες. Καμία πρόσθετη δραστικότητα κατά του όγκου δεν είχαν οι ασθενείς με τη θεραπεία συνδυασμού (72). Ο ταυτόχρονος αποκλεισμός IGF – 1R και ΜΕΚ έχει αποδειχθεί ότι αποτρέπει αποτελεσματικά την εμφάνιση των EGFR – IGF1R διασταυρούμενων επιδράσεων σε προκλινικά μοντέλα με ορθοκολικό καρκίνο και BRAF μετάλλαξη (73) .
Συμπεράσματα
Παρά την ταχεία πρόοδο στην EGFR στοχευμένη θεραπεία , πολλά μένουν ακόμη να μελετηθούν για να κατανοήσουμε τον μηχανισμό αντίστασης στον ΚΠΕΟ. Σαφώς, η KRAS μετάλλαξη στο κωδικόνιο 12 είναι η κύρια αιτία της αντίστασης στους αναστολείς του EGFR. Στην ομάδα KRAS WT, διάφοροι παράγοντες φαίνεται να επηρεάζουν την αντίσταση και αυτοί περιλαμβάνουν την έκφραση συνδέτη και την ενεργοποίηση των οδών ΡΙ3Κ ή IGFR – 1. Ο ρόλος των RAS μεταλλάξεων στο κωδικόνιο 13 και των BRAF μεταλλάξεων ως μηχανισμοί αντίστασης στους αναστολείς του EGFR απαιτεί περαιτέρω έρευνα για να επιβεβαιωθεί. Ο προσδιορισμός του μηχανισμού αντοχής στους αναστολείς του EGFR θα βελτιώσει την ικανότητά μας για την επιλογή ασθενών για εξατομικευμένη θεραπεία, καθώς και για την ανάπτυξη νέων συνδυασμένων θεραπειών που μπορούν να ξεπεράσουν την αντίσταση στις τρέχουσες διαθέσιμες θεραπείες.
Βιβλιογραφία
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012;62:10-29.
2. Centers for Disease Control and Prevention: United States Cancer Statistics: US Cancer Statistics Working Group. Available online: http://www.cdc.gov/uscs, 2013.
3. André T, Raymond E, de Gramont A. Regorafenib for metastatic colorectal cancer. Lancet 2013;381:1536-7.
4. FDA approves aflibercept (Zaltrap) for metastatic colorectal cancer. Oncology (Williston Park) 2012;26:842, 873.
5. Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007;357:2040-8.
6. Van Cutsem E, Köhne CH, Láng I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 2011;29:2011-9.
7. Aboud-Pirak E, Hurwitz E, Pirak ME, et al. Efficacy of antibodies to epidermal growth factor receptor against KB carcinoma in vitro and in nude mice. J Natl Cancer Inst 1988;80:1605-11.
8. Pollack VA, Savage DM, Baker DA, et al. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther 1999;291:739-48.
9. Sobrero AF, Maurel J, Fehrenbacher L, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:2311-9.
10. Yang XD, Jia XC, Corvalan JR, et al. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol 2001;38:17-23.
11. Borner M, Koeberle D, Von Moos R, et al. Adding cetuximab to capecitabine plus oxaliplatin (XELOX) in first-line treatment of metastatic colorectal cancer: a randomized phase II trial of the Swiss Group for Clinical Cancer Research SAKK. Ann Oncol 2008;19:1288-92.
12. Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 2010;28:4697-705.
13. Information on the conditional approval of panitumumab (Vectibix) by the EMEA. Available online: www.emea.europa.eu/pdfs/human/press/pr/32370307en.pdf2011.
14. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127-37.
15. Oda K, Matsuoka Y, Funahashi A, et al. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 2005;1:2005.0010.
16. Shaib W, Mahajan R, El-Rayes B. Markers of resistance to anti-EGFR therapy in colorectal cancer. J Gastrointest Oncol. 2013 Sep;4(3):308-18
17. Han CB, Li F, Ma JT, et al. Concordant KRAS mutations in primary and metastatic colorectal cancer tissue specimens: a meta-analysis and systematic review. Cancer Invest 2012;30:741-7.
18. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757-65.
19. Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 2007;25:1658-64.
20. Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408-17.
21. Guerrero S, Casanova I, Farré L, et al. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res 2000;60:6750-6.
22. De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010;304:1812-20.
23. Peeters M, Douillard JY, Van Cutsem E, et al. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol 2013;31:759-65.
24. Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013 Sep 12;369(11):1023-34.
25. Stintzing S, Jung A, Rossius L, et al: Analysis of KRAS/NRAS and BRAF mutations in FIRE-3. A randomized phase III study of FOLFIRI plus cetuximab or bevacizumab as first-line treatment for wild-type KRAS (exon 2) metastatic colorectal cancer patients [abstract]. ESMO European Cancer Congress 2013:E17-7073. Available at http://www.esmo.org/Conferences/Past-Conferences/European-Cancer-Congress-2013/News/Expended-Mutational-Analysis-of-Phase-III-FIRE-3-Trial-Data-Shows-Most-Patients-with-Wild-Type-RAS-Metastatic-Colorectal-Cancer-Benefit-from-First-Line-FOLFIRI-Plus-Cetuximab-Treatment
26. Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25:3230-7.
27. Baker JB, Dutta D, Watson D, et al. Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br J Cancer 2011;104:488-95.
28. Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005;23:1803-10.
29. Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 2005;6:279-86.
30. Sartore-Bianchi A, Moroni M, Veronese S, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol 2007;25:3238-45.
31. Lenz HJ, Van Cutsem E, Khambata-Ford S, et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol 2006;24:4914-21.
32. Cappuzzo F, Finocchiaro G, Rossi E, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol 2008;19:717-23.
33. Personeni N, Fieuws S, Piessevaux H, et al. Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res 2008;14:5869-76.
34. Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992-5.
35. Jiang Z, Li C, Li F, et al. EGFR gene copy number as a prognostic marker in colorectal cancer patients treated with cetuximab or panitumumab: a systematic review and meta analysis. PLoS One 2013;8:e56205.
36. De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753-62.
37. Richman SD, Seymour MT, Chambers P, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol 2009;27:5931-7.
38. Tol J, Dijkstra JR, Klomp M, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer 2010;46:1997-2009.
39. Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol 2012;30:1755-62.
40. Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008;26:5705-12.
41. Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65:6063-9.
42. Deng G, Bell I, Crawley S, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res 2004;10:191-5.
43. Wang L, Cunningham JM, Winters JL, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res 2003;63:5209-12.
44. Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100-3.
45. Ji H, Wang Z, Perera SA, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res 2007;67:4933-9.
46. Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 2009;101:715-21.
47. Vaughn CP, Zobell SD, Furtado LV, et al. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 2011;50:307-12.
48. Souglakos J, Philips J, Wang R, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 2009;101:465-72.
49. Solit D, Rosen N. Oncogenic RAF: a brief history of time. Pigment Cell Melanoma Res 2010;23:760-2.
50. Migliardi G, Sassi F, Torti D, et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res 2012;18:2515-25.
51. Mao C, Yang ZY, Hu XF, et al. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol 2012;23:1518-25.
52. Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A 2008;105:2652-7.
53. Prenen H, De Schutter J, Jacobs B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 2009;15:3184-8.
54. Pentheroudakis G, Kotoula V, De Roock W, et al. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer 2013;13:49.
55. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351-6.
56. Ihle NT, Lemos R, Jr, Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res 2009;69:143-50.
57. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010;28:1075-83.
58. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304:554.
59. Ikenoue T, Kanai F, Hikiba Y, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 2005;65:4562-7.
60. Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997;275:1943-7.
61. Guanti G, Resta N, Simone C, et al. Involvement of PTEN mutations in the genetic pathways of colorectal cancerogenesis. Hum Mol Genet 2000;9:283-7.
62. Shin KH, Park YJ, Park JG. PTEN gene mutations in colorectal cancers displaying microsatellite instability. Cancer Lett 2001;174:189-94.
63. Goel A, Arnold CN, Niedzwiecki D, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 2004;64:3014-21.
64. Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009;69:1851-7.
65. Barault L, Veyrie N, Jooste V, et al. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. Int J Cancer 2008;122:2255-9.
66. Slattery ML, Lundgreen A, Wolff RK. MAP kinase genes and colon and rectal cancer. Carcinogenesis 2012;33:2398-408.
67. Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011;3:99ra86.
68. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486:532-6.
69. Reinmuth N, Liu W, Fan F, et al. Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res 2002;8:3259-69.
70. Hu YP, Patil SB, Panasiewicz M, et al. Heterogeneity of receptor function in colon carcinoma cells determined by cross-talk between type I insulin-like growth factor receptor and epidermal growth factor receptor. Cancer Res 2008;68:8004-13.
71. Bohula EA, Playford MP, Macaulay VM. Targeting the type 1 insulin-like growth factor receptor as anti-cancer treatment. Anticancer Drugs 2003;14:669-82.
72. Reidy DL, Vakiani E, Fakih MG, et al. Randomized, phase II study of the insulin-like growth factor-1 receptor inhibitor IMC-A12, with or without cetuximab, in patients with cetuximab- or panitumumab-refractory metastatic colorectal cancer. J Clin Oncol 2010;28:4240-6.
73. Buck E, Eyzaguirre A, Rosenfeld-Franklin M, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res 2008;68:8322-32.






