
Από τους
Βασίλειο Ραμφίδη και Νικόλαο Κεντεποζίδη
Παθολόγους Ογκολόγους
Ο καρκίνος του πνεύμονα αποτελεί την πρωτεύουσα αιτία θνησιμότητας παγκοσμίως. Περίπου 230000 νέες περιπτώσεις καρκίνου του πνεύμονα εκτιμάται ότι διαγιγνώσκονται ετησίως μόνο στις Ηνωμένες Πολιτείες Αμερικής και οι αναμενόμενοι θάνατοι είναι περί των 160000. Παρά την πρόοδο στην έρευνα του καρκίνου, η πενταετής επιβίωση παραμένει φτωχή και αγγίζει το 15%. Με μια ιστορική ματιά, ο μη μικροκυτταρικός καρκίνος του πνεύμονα (NSCLC) αντιμετωπιζόταν σαν μία ενιαία νοσολογική οντότητα και η χημειοθεραπεία στο μεταστατικό καρκίνο είχε περορισμένα αποτελέσματα όσων αφορά τη συνολική επιβίωση και την εξασφάλιση ικανοποιητικής ποιότητας ζωής. Μια σειρά από μεγάλες τυχαιοποιημένες φάσης ΙΙΙ κλινικές μελέτες καθιέρωσαν τη χημειοθεραπεία με διπλέτα βασισμένη στην πλατίνα ως standard of care στη θεραπεία του μεταστατικού μη μικροκυτταρικού καρκίνου του πνεύμονα με ποσοστά ανταπόκρισης 20 με 30 % και μέση επιβίωση 8 με 11 μήνες. Αυτή η θεραπευτική προσέγγιση φαίνεται να έχει φτάσει σε ένα plateau αποτελεσματικότητας και αυτό οδήγησε σε νέες εξελίξεις στη διαχείρηση αυτών των ασθενών.
Πρόσφατες μελέτες στην αντιμετώπιση του μεταστατικού μη μικροκυτταρικού καρκίνου του πνεύμονα (NSCLC) οδήγησαν στη διαπίστωση ότι ο NSCLC δεν αποτελεί αποκλειστικά μία νοσολογική οντότητα, αλλά διαφορετικά νεοπλάσματα καθοδηγούμενα από διαφορετικά μοριακά μονοπάτια, διαφορετική βιολογική συμπεριφορά και κατ’ επέκταση χρήζουν διαφορετική αντιμετώπιση (εικόνα 1). Ο Lynch και συνεργάτες και ο Paez και συνεργάτες ήταν οι πρώτοι που περιέγραψαν ένα υποσύνολο ασθενών με μη μικροκυτταρικό καρκίνο του πνεύμονα που εμφανίζουν μεταλλάξεις του EGFR γονιδίου και οι οποίοι ανταποκρίθηκαν σε θεραπεία με μικρομοριακούς αναστολείς της τυροσινικής κινάσης (TKIs). Η ανακάλυψη αυτή αποτέλεσε δια παντός τη θεραπευτική προσέγγιση του μη μικροκυτταρικού καρκίνο του πνεύμονα και η νέα κατεύθυνση είναι πλέον μια πιο εξατομικευμένη προσέγγιση βασισμένη στα ενεργοποιημένα μοριακά μονοπάτια του όγκου. Μια προσέγγιση που δεν αφορά μόνο τις θεραπείες που στοχεύουν EGFR υποδοχείς σε αδενοκαρκίνωμα πνεύμονα με EGFR μεταλλάξεις, αλλά και σε ALK μεταλλάξεις πνεύμονα και πιο πρόσφατα σε περιπτώσεις αδενοκαρκίνωμα πνεύμονα που εκφράζουν ROS1 και BRAF μεταλλάξεις.
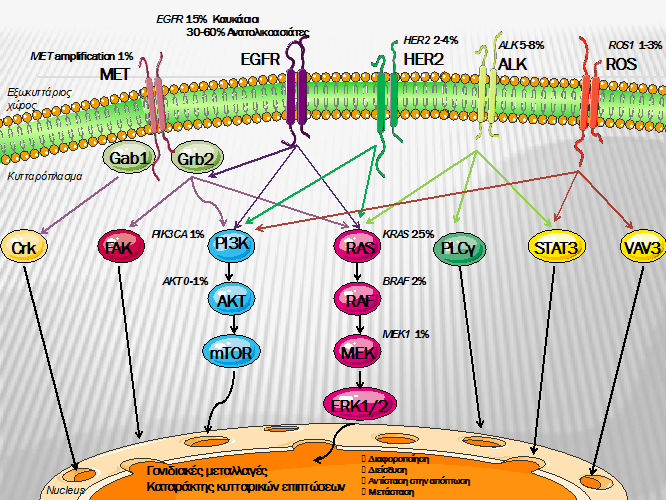
Εικόνα 1: Κυτταρικά μονοπάτια στο μη μικροκυτταρικό καρκίνο του πνεύμονα και η συχνότητα των οδηγών μεταλλάξεων στο αδενοκαρκίνωμα πνεύμονα
EGFR μονοπάτι στον καρκίνο του πνεύμονα
Το EGFR είναι μέλος της οικογένιεας των ErbB (ή HER) υποδοχέων της τυροσινικής κινάσης και αποτελεί καταλυτικό κρίκο στην ενεργοποίηση κυτταρικών μοριακών μονοπατιών όπως το RAS-RAF-MEK μονοπάτι και το PI3K-AKT-mTOR μονοπάτι, αλλά και δύναται να επηρεαστούν και από άλλους υποδοχείς τυροσινικής κινάσης όπως ο insulin-like growth factor υποδοχέας (IGF1-R) και ΜΕΤ. Τα μονοπάτια αυτά επηρεάζουν την κυτταρική διαφοροποίηση του καρκινικού κυττάρου, την τοπική επέκταση, τη μεταστατική του ικανότητα, την αντίσταση στην απόπτωση και την αγγειογένεση. Οι EGFR μεταλλάξεις πιο συχνά λαμβάνουν χώρα με έλλειψη του εξωνίου 19 (45%) και αντικατάσταση του L858R του εξωνίου 21 (40-45 %) και πιο σπάνια με αντικατάσταση νουκλεοτιδίων στο εξώνιο 18 και προσθήκη στο εξώνιο 20 σε ποσοστό 5 %. Οι EGFR μεταλλάξεις είναι παρούσες στο 15 % των βόρειων αμερικάνων και δυτικοευρωπαίων με καρκίνο του πνεύμονα, 30 με 50 % ανατολικών ασιατών, ενώ το ποσοστό ξεπερνάει το 50 % των ασιατών που δεν κάπνισαν ποτέ με ιστολογική αδενοκαρκινώματος.
Η αναγνώριση του βασικού ρόλου των στοχεύουσων θεραπειών σε ασθενείς αδενοκαρκίνωμα πνεύμονα και EGFR μεταλλάξεις επιβεβαιώθηκε από τη μελέτη IPASS (Iressa Pan-Asia Study), μια τυχαιοποιημένη φάσης ΙΙΙ κλινική μελέτη που είχε ως στόχο την εκτίμηση της αποτελεσματικότητας του gefitinib σε σύγκριση με carboplatin και paclitaxel στην πρώτη γραμμή θεραπείας σε επιλεγμένους ασθενείς με ιστολογική αδενοκαρκινώματος, ασιατική εθνότητα που δεν είχαν καπνίσει ποτέ ή ήταν ελαφρείς καπνιστές. Η μελέτη IPASS αποτέλεσε την πρώτη μελέτη που φανέρωσε την υπεροχή του gefitinib, ενός μικρομοριακού αναστολέα του EGFR, έναντι της μέχρι πρότεινος καθιερωμένη θεραπεία βασισμένη στην πλατίνα σε ασθενείς με καρκίνο του πνεύμονα και μεταλλάξεις του EGFR. Η υπεροχή αυτή εκφράστηκε με 71,2% ποσοστό ανταποκρίσης (response rate / RR) και 52 % μείωση του σχετικού κινδύνου υποτροπής (progression-free survival / PFS) συγκρινόμενο με τη χημειοθεραπεία (p<0.001) (Πίνακας 1). Αντίθετα, ασθενείς που δεν είχαν μετάλλαξη του EGFR και έλαβαν gefitinib εμφάνισαν χειρότερα ποσοστά ανταπόκρισης (RR) (1.1% προς 23.5% αντίστοιχα) και μικρότερο διάστημα ελεύθερο νόσου (PFS) (HR 2.85, p<0.001; πίνακας 1). Συνεπώς, είναι αναποτελεσματικό να αντιμετωπίζεις ασθενείς με EGFR μικρομοριακούς αναστολείς χωρίς να έχει προηγηθεί μοριακός έλεγχος μεταλλάξεων και θα πρέπει όλοι οι ασθενείς με αδενοκαρκίνωμα πνεύμονα να ελέγχεται το μοριακό τους προφίλ.
Πίνακας 1. Κλινικές μελέτες μη μικροκυτταρικού καρκίνου πνεύμονα με EGFR μεταλλάξεις |
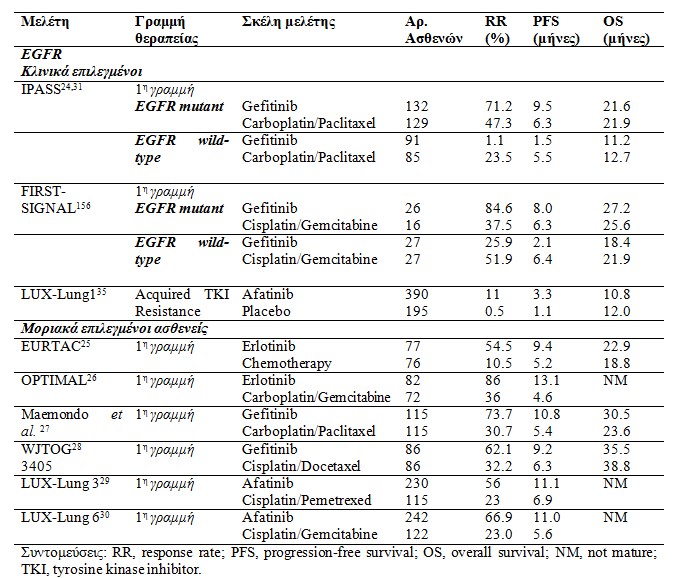
Το όφελος από τη χρήση μικρομοριακών αναστολέων σε ασθενείς με EGFR μεταλλάξεις επιβεβαιώθηκε από έξι τυχαιοποιημένες μελέτες φάσης ΙΙΙ που διερεύνησαν το ρόλο των gefitinib, erlotinib και afatinib. Στις μελέτες αυτές τα ποσοστά ανταπόκρισης (RRs) κυμαίνονταν από 55 έως 86% και συχετίζονταν με αξιοσημείωτο διάστημα PFS (Progression Free Survival) (πίνακας 1). Πρέπεί να αναφερθεί ότι τόσο η μελέτη IPASS, όσο και οι μελέτες από τον Rossell (EURTAC), τον Maemondo και Mitsudomi (WJTOG 3405) δεν ανέδειξαν στατιστικά σημαντικό όφελος για τους ασθενείς που έλαβαν EGFR μικρομοριακούς αναστολείς κι αυτό διότι ο σχεδιασμός των μελετών προέβλεπε crossover των ασθενών που έλαβαν χημειοθεραπεία, ώστε σε περίπτωση υποτροπής να λάβουν κι αυτοί EGFR TKIs. Τα ποσοστά των crossover ήταν παρόμοια. 73% για τη μελέτη EURTAC, 95% στη μελέτη από τον Maemondo και των συνεργατών και 91% στη μελέτη WJTOG.
Όπως διαφαίνεται από τις παραπάνω μελέτες η χορήγηση μικρομοριακών αναστολέων EGFR ως πρώτη γραμμή σε σύγκριση με δεύτερη γραμμή δεν επηρεάζει στατιστικώς σημαντικά τη συνολική επιβίωση (OS). Πρέπει σε αυτό το σημείο να σημειωθεί ότι ένα μικρό ποσοστό ασθενών με EGFR μεταλλάξεις που έλαβαν αρχικά χημειοθεραπεία δε μπόρεσαν τελικά να λάβουν EGFR TKIs λόγω ταχείας απιδείνωσης. Αυτοί οι ασθενείς ενδεχομένως να οφελούνταν αν λάμβαναν ως πρώτη γραμμή TKI.
Μηχανισμοί αντίστασης στους μικρομοριακούς αναστολείς EGFR μεταλλάξεων στον καρκίνο του πνεύμονα
Ίσως το πιο καταλυτικό σημείο όσων αφορά τις στοχεύουσες θεραπείες και τους ασθενείς με EGFR μεταλλάξεις και αδενοκαρκίνωμα του πνεύμονα είναι η ανάπτυξη μηχανισμών αντίστασης στους μικρομοριακούς αναστολείς (σχήμα 2).
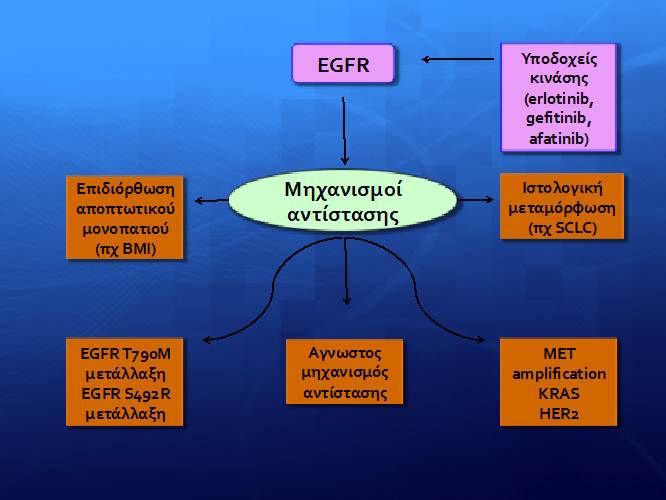
Σχήμα 2: Μηχανισμοί αντίστασης στους μικρομοριακούς αναστολείς EGFR
Πρωτοπαθής αντίσταση (de novo)
Παρά το σημαντικό κλινικό όφελος που προκύπτει από τη χορήγηση μικρομοριακών αναστολέων σε EGFR μεταλλάξεις υπάρχουν ένα ποσοστό ασθενών που φέρουν μετάλλαξη αλλά δεν οφελούνται από EGFR στοχεύουσες θεραπείες με το όφελος στο μέσο διάστημα PFS να είναι απογοητευτικό σε σύγκριση με τη καθιερωμένη χημειοθεραπεία. Υπάρχουν δύο βασικοί μηχανισμοί πρωτοπαθούς αντίστασης: (1) δευτεροπαθείς μεταλλαγές του EGFR που εμποδίζουν την αναστολή του υποδοχέα EGFR από EGFR μικρομοριακό ανστολέα και (2) επιπρόσθετες γενετικές μεταλλαγές που συνυπάρχουν με τη μετάλλαξη EGFR σε ασθενείς με μη μικροκυτταρικό καρκίνο πνεύμονα (πίνακας 2).
Γύρω στο 5% των ασθενών με αδενοκαρκίνωμα πνεύμονα και EGFR μεταλλάξεις εμφανίζει προσθήκη (insertion) του εξωνίου 20 η οποία είναι λιγότερη ευαίσθητη στη θεραπεία με EGFR μικρομοριακών αναστολέων. Επιπρόσθετα, ασθενείς μςμετάλλαξη EGFR T790M στο εξώνιο 20 εμφανίζουν επίσης αντίσταση στη θεραπεία με EGFR TKIs. Σε προθεραπευμένους ασθενείς με T790M μετάλλαξη, χαμηλή έκφραση του BRCA1 mRNA σχετίζεται με μεγαλύτερο PFS κατά τη χορήγηση erlotinib. Τα δεδομένα προτείνουν ότι χαμηλά επίπεδα BRCA1 ενδεχομένως να αναιρούν τη αρνητική επιρροή της EGFR T790M μετάλλαξης, ενώ υψηλά επίπεδα έκφρασης BRCA1 να οδηγούν σε de novo ανάπτυξη αντίστασης. Επιπλέον της EGFR T790M, δευτεροπαθείς EGFR μεταλλάξεις δύναται να προκαλέσουν πρωτοπαθή αντίσταση (πχ D761Y και L858R).
Η παρουσία γενετικών μεταλλαγών είναι δυνατόν να ενοχοποιηθεί επίσης για τη de novo αντίσταση. Η ενεργοποίηση του μονοπατιού PI3CA/AKT όπως προκύπτει μετά από απώλεια έκφρασης PTEN μειώνει την ευαισθησία στους EGFR TKIs. Κυτταρικές σειρές σε προκλινικές μελέτες έδειξαν επίσης ότι το IGF1R (insulin growth factor 1 receptor) μονοπάτι μπορεί να επηρεάζει τη αντίσταση. Τέλος ενεργοποίηση του NFκB σήματος ενδεχομένως να αποτελεί έναν επιπλέον μηχανισμό αντίστασης στους EGFR ΤΚΙς όπως προτείνουν ορισμένες μελέτες.
|
Μικρομοριακοί αναστολείς EGFR στον καρκίνο του πνεύμονα |
|
|
Πρωτοπαθής αντίσταση (de novo) |
EGFR exon 20 insertion |
BIM deletion
EGFR T790M
Ενεργοποίηση PI3CA/AKT και IGF1R μονοπατιού
Πίνακας 2: Κύριοι μηχανισμοί πρωτοπαθούς (de novo) αντίστασης
Επίκτητη αντίσταση (acquired resistance)
Ο πιο συχνός επίκτητος μηχανισμός αντίστασης στους μικρομοριακούς αναστολείς EGFR σε ασθενείς με καρκίνο του πνεύμονα και EGFR μεταλλάξεις είναι η δευτερογενής μετάλλαξη T790M, η οποία ανιχνεύται στο 60% των ασθενών που εμφανίζουν επίκτητη αντίσταση. Οι δευτερογενείς μεταλλάξεις αποτελούν έναν πολύ συχνό μηχανισμό αντίστασης. Παρόμοιο παράδειγμα αποτελεί η ABL T315I στη χρόνια μυελογενή λευχαιμία (ΧΜΛ), η KIT T670I στους GIST (gastrointestinal stromal tumors) όγκους και η ALK L 1196M στα αδενοκαρκινώματα. Τόσο η μετάλλαξη T790M, όσο και η ALK F1147L έχουν τον ίδιο μηχανισμό δράσης αυξάνοντας την ανάγκη των κινασών για ATP κατά πέντε φορές, με αποτέλεσμα να μειώνεται η ευαισθησία των υποδοχέων erlotinib και crizotinib αντίστοιχα.
Σε αντίθεση με τα κυτταροτοξικά χημειοθεραπευτικά σκευάσματα η αντίσταση στις αντί-EGFR στοχεύουσες θεραπείες στην ενεργοποίηση και αλληλεπίδραση πολλαπλών μοριακών μονοπατιών (σχήμα 1). Τα μονοπάτια αυτά αποτελούν ένα πολύπλοκο δίκτυο που καθορίζουν την ανάπτυξη του κυττάρου, την επιβίωσή του, τη διαφοροποίησή του και την αποπτωτική του ικανότητα. Η ενεργοποίηση κάποιων εξ αυτών των μονοπατιών μπορούν να αποτελέσουν μηχανισμούς αντίστασης. Η ενίσχυση του MET μονοπατιού (MET amplification) παρατηρείται στο 5 με 20% των ασθενών, ενίσχυση του HER2 μονοπατιού (HER2 amplification) στο 12% των ασθενών και BRAF μεταλλάξεις στο 1% των ασθενών.
Η μετάπτωση του αδενοκαρκινώνατος σε μικροκυτταρικό κακρίνωμα η οποία παρατηρήθηκε πρώτη φορά το 2010 αποτελεί έναν επιπρόσθετο μηχανισμό αντίστασης. Η ιστολογική αυτή μετάπτωση έχει παρατηρηθεί στο 3 με 15% των περιπτώσεων και ασθενείς αυτοί μπορούν να ευνοηθούν από τη χορήγηση πλατίνας και etoposide εφόσον επιβεβαιωθεί η αλλαγή αυτή με νέα ιστολογική βιοψία.
|
Μικρομοριακοί αναστολείς EGFR στον καρκίνο του πνεύμονα |
|
|
Επίκτητη αντίσταση (Acquired resistance) |
T790M μετάλλαξη (60%) |
MET amplification (5-20%)
HER2 amplification (12%)
BRAF μετάλλαξη (1%)
Μετάπτωση σε SCLC (3-15%)
Άγνωστος μηχανισμός (30%)
Πίνακας 3: Κύριοι μηχανισμοί επίκτητης (acquired) αντίστασης
Στρατηγικές παράκαμψης των μηχανισμών αντίστασης
Η αναγνώριση των μηχανισμών αντίστασης αποτελεί το πρώτο βήμα για την ανάπτυξη στρατηγικών παράκαμψης. Οι στρατηγικές αυτές περιλαμβάνουν τόσο νέας γενιάς EGFR αναστολείς, όσο και συνδυασμό στοχευουσών θεραπειών.
Νέας γενιάς μικρομοριακοί αναστολείς
Το 2005, οι νέας γενιάς μικρομοριακοί αναστολείς δοκιμάστηκαν σε EGFR T790M μεταλλάξεις σε προ-κλινικά μοντέλα κυτταρικών σειρών με αδενοκαρκίνωμα πνεύμονα. Το επόμενο βήμα επιτεύχθει το 2009 με αναστολείς που στοχεύουν ειδικά σε T790M EGFR μεταλλάξεις. Τέτοιοι αναστολείς που πλέον δοκιμάζονται σε κλινικές μελέτες είναι το afatinib (BIBW 2992) και το dacomitinib (PF00299804) οι οποίοι και οι δύο αναστέλουν τη δράση και του HER2 και του HER4 και ο CO-1686. Ο AZD9291 έδειξε ελπιδοφόρα αποτελέσματα σε φάσης Ι μελέτη, ενώ η ανάπτυξη των neratinib (HKI-272) και canertinib (CI-1033) διακόπηκε έλλειψης αποτελεσματικότητας και μη ανεκτής τοξικότητας. Ο αναστολέας midostaurin (PKC412) ένας αναστολέας των πρωτείνης κινάσης C (PCK), FLT και KIT που για την ώρα δοκιμάζεται στην οξεία μυεολογενή λευχαιμία φάνηκε ότι μπορεί να στοχεύσει την T790M μετάλλαξη. Ο AP26113, ένας ALK υποδοχέας φάνηκε επίσης ότι δρα ανασταλτικά επί του EGFR T790M. Στη LUX-Lung 1 κλινική μελέτη αξιολογήθηκε το afatinib σε ασθενείς που εμφάνισαν επιδείνωση μετά από χορήγηση erlotinib ή gefitinib. Παρόλο που αυτή η μελέτη δε φανέρωσε σημαντικό πλεονέκτημα όσων αφορά τη συνολική επιβίωση συγκρινόμενη με best supportive care (HR 1.08, P=0.74), υπήρχε σημαντικό όφελος στο PFS στο σκέλος του afatinib (HR 0.38, P<0.0001).
Συνδυασμός θεραπειών
Πολλές τρέχουσες κλινικές μελέτες περιλαμβάνουν συνδυασμό πολλών σκευασμάτων που στοχεύουν στο MET ή HGF (hepatocyte growth factor), το dasatinib, everolimus, bortezomib, bevacizumab, sunitinb και cetuximab. Μέχρι στιγμής ωστόσο μόνο ο συνδυασμός afatinib και cetuximab φαίνεται να προσφέρει κλινικό όφελος στον καρκίνο του πνεύμονα. Ένας πιθανός λόγος που δεν έχουν ανευρεθεί ακόμα αποτελεσματικοί συνδυασμοί είναι ότι δεν υπάρχουν πολλές κλινικές μελέτες που να απευθύνονται σε ασθενείς με επιβεβαιωμένη αντίσταση στους μικρομοριακούς αναστολείς. Γι’ αυτό και η βιοψία μετά από χορήγηση μικρομοριακών αναστολέων του EGFR και μετά από υποτροπή, είναι απαραίτητη προκειμένου να αναπτυχθούν αποτελεσματικές νέες θεραπευτικές στρατηγικές σε ασθενείς με αντίσταση στους EGFR TKIs.
Βιβλιογραφία
1. Howlader N, Noone AM, Krapcho M, et al. SEER Stat Fact Sheets: Lung and Bronchus. http://seercancergov/csr/1975_2008/ 2010.
2. Hopwood P, Stephens RJ. Symptoms at presentation for treatment in patients with lung cancer: implications for the evaluation of palliative treatment. The Medical Research Council (MRC) Lung Cancer Working Party. Br J Cancer 1995;71:633-6.
3. Burdett S, Stephens R, Stewart L, et al. Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: a systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J Clin Oncol 2008;26:4617-25.
4. Burdett S, Stewart L, Pignon J-P. Chemotherapy in non-small cell lung cancer: An update of an individual patient data-based meta-analysis. J Thorac Cardiovasc Surg 2005;129:1205-.
5. Rapp E, Pater JL, Willan A, et al. Chemotherapy can prolong survival in patients with advanced non-small-cell lung cancer–report of a Canadian multicenter randomized trial. J Clin Oncol 1988;6:633-41.
6. Non-small Cell Lung Cancer Collaborative Group. Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. Br Med J 1995;311:899-909.
7. Fossella F, Pereira JR, von Pawel J, et al. Randomized, multinational, phase III study of docetaxel plus platinum combinations versus vinorelbine plus cisplatin for advanced non-small-cell lung cancer: the TAX 326 study group. J Clin Oncol 2003;21:3016-24.
8. Kelly K, Crowley J, Bunn PA, Jr., et al. Randomized phase III trial of paclitaxel plus carboplatin versus vinorelbine plus cisplatin in the treatment of patients with advanced non–small-cell lung cancer: a Southwest Oncology Group trial. J Clin Oncol 2001;19:3210-8.
9. Scagliotti GV, De Marinis F, Rinaldi M, et al. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol 2002;20:4285-91.
10. Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 2002;346:92-8.
11. Zatloukal P, Petruzelka L, Zemanova M, et al. Gemcitabine plus cisplatin vs. gemcitabine plus carboplatin in stage IIIb and IV non-small cell lung cancer: a phase III randomized trial. Lung Cancer 2003;41:321-31.
12. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39.
13. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500.
14. Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703.
15. Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863-70.
16. Ou SI, Bang Y, Camidge DR, et al. Efficacy and safety of crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer (NSCLC). J Clin Oncol 2013;Suppl:abstr 8032.
17. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367-80.
18. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007;7:169-81.
19. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57.
20. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-46.
21. Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735-42.
22. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8.
23. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121-8.
24. Yang JC-H, Schuler MH, Yamamoto N, et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol 2012;30:LBA7500.
25. Wu YL, Zhou C, Hu C, et al. LUX-Lung 6: A randomized, open-label, phase III study of afatinab (A) versus gemcitabine/cisplatin (GC) as first-line treatment for Asian patients (pts) with EGFR mutation-positive (EGFR M+) advanced adenocarcinoma of the lung. J Clin Oncol 2013;31:abstr 8016.
26. Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol 2011;29:2866-74.
27. Mok T, Yang JJ, Lam KC. Treating patients with EGFR-sensitizing mutations: first line or second line–is there a difference? J Clin Oncol 2013;31:1081-8.
28. J. Y. Wu, S. G. Wu, C. H. Yang et al., “Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response,” Clinical Cancer Research, vol. 14, no. 15, pp. 4877–4882, 2008.
29. S. Maheswaran, L. V. Sequist, S. Nagrath et al., “Detection of mutations in EGFR in circulating lung-cancer cells,” New England Journal of Medicine, vol. 359, no. 4, pp. 366–377, 2008.
30. M. Inukai, S. Toyooka, S. Ito et al., “Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer,” Cancer Research, vol. 66, no. 16, pp. 7854–7858, 2006.
31. L. Prudkin, X. Tang, and I. I. Wistuba, “Germ-line and somatic presentations of the EGFR T790M mutation in lung cancer,” Journal of Thoracic Oncology, vol. 4, no. 1, pp. 139–141, 2009.
32. R. Rosell, M. A. Molina, C. Costa et al., “Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations,” Clinical Cancer Research, vol. 17, no. 5, pp. 1160–1168, 2011.
33. M. L. Sos, M. Koker, B. A. Weir et al., “PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of akt and EGFR,” Cancer Research, vol. 69, no. 8, pp. 3256–3261, 2009.
34. I. Vivanco, D. Rohle, M. Versele et al., “The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation,” Proceedings of the National Academy of Sciences of the United States of America, vol. 107, no. 14, pp. 6459–6464, 2010.
35. Y. Ben-Neriah and M. Karin, “Inflammation meets cancer, with NF-κB as the matchmaker,” Nature Immunology, vol. 12, no. 8, pp. 715–723, 2011.
36. T. G. Bivona, H. Hieronymus, J. Parker et al., “FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR,” Nature, vol. 471, no. 7339, pp. 523–526, 2011.
37. Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011;17:1169-80.
38. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240-7.
39. Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2012;2:922-33.
40. Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 2012;109:E2127-33.
41. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
42. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007;104:20932-7.
43. Engelman JA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43.
44. Bridges, A.J. The rationale and strategy used to develop a series of highly potent, irreversible, inhibitors of the epidermal growth factor receptor family of tyrosine kinases. Curr. Med. Chem. 6, 825–843 (1999).
45. Kwak, E. The role of irreversible HER family inhibition in the treatment of patients with non-small cell lung cancer. Oncologist 16, 1498–1507 (2011).
46. Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005).
47. Kwak, E.L. et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 102, 7665–7670 (2005).
48. Zhou, W. et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009).
49. Dienstmann, R., De Dosso, S., Felip, E. & Tabernero, J. Drug development to overcome resistance to EGFR inhibitors in lung and colorectal cancer. Mol. Oncol. 6, 15–26 (2012).
50. Yu, H.A. & Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Canc. Netw. 11, 161–169 (2013).
51. Ranson, M. et al. Preliminary results from a Phase I study with AZD9291: an irreversible inhibitor of epidermal growth factor receptor (EGFR) activating and resistance mutations in non-small cell lung cancer (NSCLC). European Cancer Conference (Amsterdam, 2013).
52. Ou, S.H. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Crit. Rev. Oncol. Hematol. 83, 407–421 (2012).
53. Lee, H.J. et al. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov. 3, 168–181 (2013).
54. Rivera, V.M. et al. AP26113 is a dual ALK/EGFR inhibitor: characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res. 72 (suppl. 1), abstract 1794 (2012).
55. Janjigian, Y. et al. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J. Clin. Oncol. 29, 7525 (2011).
56. Janjigian, Y.Y. et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res. 17, 2521–2527 (2011).
57. Robinson, K.W. & Sandler, A.B. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist 18, 115–122 (2013).
58. Johnson, M.L. et al. Phase II trial of dasatinib for patients with acquired resistance to treatment with the epidermal growth factor receptor tyrosine kinase inhibitors erlotinib or gefitinib. J. Thorac. Oncol. 6, 1128–1131 (2011).
59. Riely, G.J. et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin. Cancer Res. 13, 5150–5155 (2007).
60. Lynch, T.J. et al. A randomized phase 2 study of erlotinib alone and in combination with bortezomib in previously treated advanced non-small cell lung cancer. J. Thorac. Oncol. 4, 1002–1009 (2009).
61. Herbst, R.S. et al. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet 377, 1846–1854 (2011).
62. Scagliotti, G.V. et al. Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J. Clin. Oncol. 30, 2070–2078 (2012).






